Синтез пиррольных интермедиатов для высокосопряженных порфиринов
МИНИСТЕРСТВО ОБРАЗОВАНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
МОСКОВСКАЯ ГОСУДАРСТВЕННАЯ АКАДЕМИЯ ТОНКОЙ
ХИМИЧЕСКОЙ ТЕХНОЛОГИИ им. М.В. Ломоносова
Кафедра химии и технологии тонких
органических соединений
МАГИСТЕРСКАЯ ДИССЕРТАЦИЯ
на тему: “СИНТЕЗ ПИРРОЛЬНЫХ ИНТЕРМЕДИАТОВ ДЛЯ ВЫСОКО СОПРЯЖЕННЫХ ПОРФИРИНОВ”
Зав.каф.ХТТОС,
акад. РИА, проф. Миронов А.Ф.
Руководитель
к.х.н., доцент Харитонова О.В.
Диссертант Ефимкин А.Г.
Москва 2002
Содержание.
1. Введение………………………………………………………………………..3-4
2. Литературный обзор………………………………………………………….5-30
2.1. Синтез замещенных пирролов……………………………………………..5-17
2.1.1. Образование связей C–N и С–С в результате реакции аминогруппы и метиленовой группы с карбонильной……………………………………………5-9
2.1.2. Конденсации, при которых в готовый углеродный скелет вводится
атом азота при помощи аммиака или аминов………………………………….9-11
2.1.3. Реакции присоединения аминов по кратным связям………………….11-14
2.1.4. Реакции образования циклов в результате внутримолекулярной конденсации……………………………………………………..……………..14-17
Методы синтеза порфиринов…………………………………………….18-30
Метод Адлера-Лонго…………………………………………………...18-21
“2+2” порфириновый синтез…………………………………………...22-28
2.2.2.1 Синтез порфиринов через b-билены…………………………………..26-27
2.2.2.2. Циклизация а,с-биладиенов…………………………………………...27-28
“3+1” порфириновый синтез…………………………………………...28-30
Обсуждение результатов………………………………………………….31-41
Охрана труда……………………………………………………………….42-53
Экспериментальная часть…………………………………………………54-67
Выводы…………………………………………………………………………68
Литература…………………………………………………………………69-76
Введение.
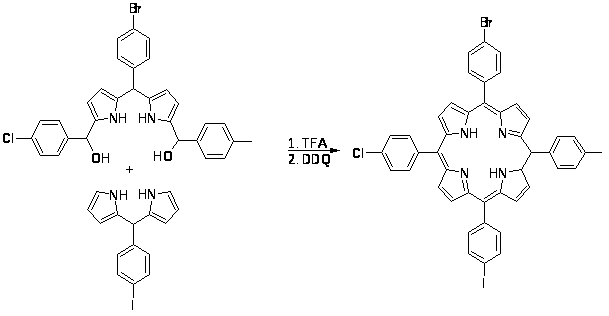
Исторически большинство исследований в области химии порфиринов были направлены на изучение, синтез и определение биохимических свойств природных тетрапиррольных пигментов (гемов, хлорофиллов, и т.д.), но в последние 20 лет наблюдается перенаправление усилий для синтеза нестандартных порфириновых систем и использования их в новых материалах. Например, порфиринсодержащие системы исследуются на возможность применения в качестве молекулярных механизмов, молекулярных проводников и молекулярных фотонных проводников. Порфирины и структурно родственные им фталоцианины всесторонне исследуются на возможность их применения в жидкокристаллических мониторах и светопоглощающих материалах.
Большинство исследований порфиринов ранее проводилось с использованием мезо-тетраарилпорфиринов, частично из-за того, что эти системы могут быть легко получены по известным литературным методам. Модифицированные системы, которые имеют хромофорные системы, поглощающие в красной и инфракрасной областях спектра, имеют огромное значение и это стимулирует дальнейшие исследования порфириновых аналогов, включающих фуран- и тиофенсодержащие макроциклы, структурные изомеры порфиринов, и так называемые расширенные порфирины. Порфирины, расширенные ароматическими фрагментами, могут выступать в качестве высоко модифицированных хромофоров, при этом сохраняя свойства, связанные с порфириновым ядром. [1]
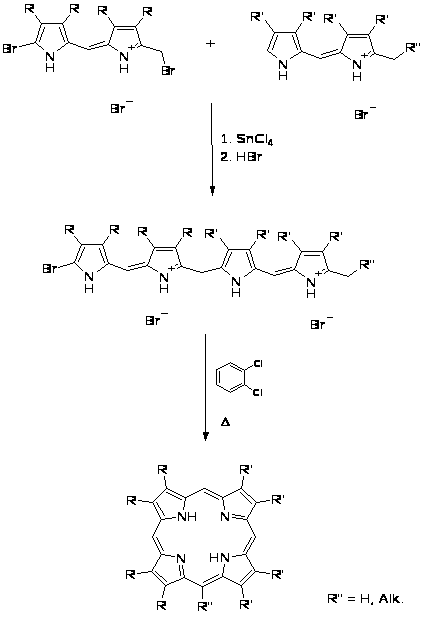
Расширение электронной порфиринового макроцикла при введение бензольного кольца приводит к значительному сдвигу в красную область спектра и уменьшает окислительный потенциал этих соединений. Высоко сопряженные порфирины, например бензопорфирины (1) и пиридинопорфирины (2), проявляют некоторые свойства, которые могут быть использованы в области медицины и новых материалов для электронной техники. [2,3].
Новым этапом в химии высоко сопряженных порфиринов явилась дальнейшая модификация хромофорной системы порфиринового ядра путем введения более сложных ароматических и гетероароматических систем. На этом этапе наряду с традиционным методом расширения порфиринового макроцикла – наращиванием ароматического ядра на основе функционально замещенного порфирина [2,3,4], был разработан новый более продуктивный метод, заключающийся в формировании сопряженной по -положениям пиррольного кольца системы пиррола и ароматического фрагмента, с последующим введением этой системы в порфириновую конденсацию [5,6,7]. На основе последнего подхода были синтезированы фенантропорфирины (3) и фенантролинопорфирины (4), а также тиадиазолопорфирины (5), которые нашли применение в качестве молекулярных зондов, фотосенсоров в фотодинамической терапии рака, геохимических стандартов при анализе осадочных металлопорфиринов [7].
Данная работа посвящена синтезу пиррольных интермедиатов для высоко сопряженных порфиринов, которые являются ключевым звеном в реализации последнего подхода.
Литературный обзор.
Синтез замещенных пирролов.
Для синтеза пиррольного цикла предложено большое количество методов. В этой работе будут рассмотрены лишь некоторые из них.
2.1.1. Образование связей C–N и С–С в результате реакции аминогруппы и метиленовой группы с карбонильной.
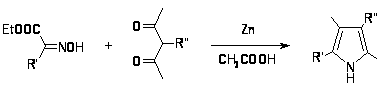
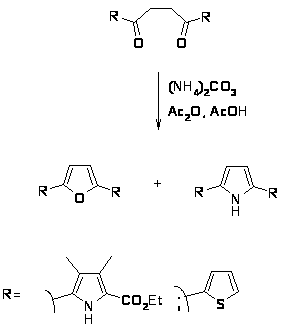
Синтез Кнорра наиболее общий и широко используемый метод получения пирролов [8]. Он заключается в конденсации -аминокетонов и -амино--кетоэфиров с кетонами или кетоэфирами в присутствии уксусной кислоты и реже щелочи. Реакции обычно протекают с хорошим выходом. -Аминокетоны получают восстановлением цинком в уксусной кислоте из предварительно полученных изонитрозо--кетоэфиров или изонитрозо- дикетонов[9,10].
Модификацией этого метода является конденсация предварительно приготовленной соли аминокетона с кетоэфиром в щелочной среде [11]. При этом получаются пирролы, содержащие в положениях 2 и 3 метильные группы, а в положениях 4 и 5 электроноакцепторные заместители. Кроме указанного восстановителя успешно был применен дитионит натрия [12].
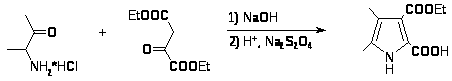
В качестве изонитрозокетонов и изонитрозокетоэфиров могут быть использованы нитрозомалоновый или нитрозоциануксусный эфиры, которые после восстановления в условиях конденсации Кнорра реагируют с -дикетонами [13].
Синтез пирролов по Кнорру удобнее проводить без выделения промежуточного аминокетона [14]. Исследование этой реакции показало, что основным ограничением при использовании этой реакции является склонность -аминокетонов к димеризации [15].
Механизм реакции Кнорра можно представить следующим образом:
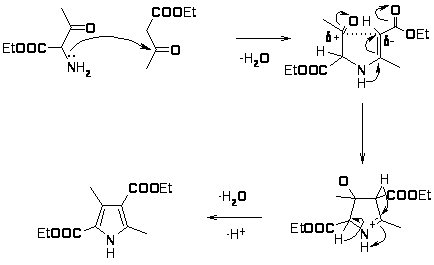
По этому же механизму протекает синтез пирролов по Кнорру в модификации Трейбса, при этом сначала происходит восстановление гидразона -дикетона до гидразина, а затем образование енамина и альдольная конденсация протекающая с хорошими выходами 60 70% [16].
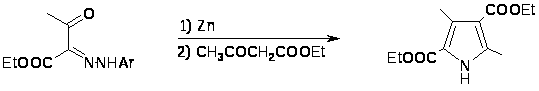
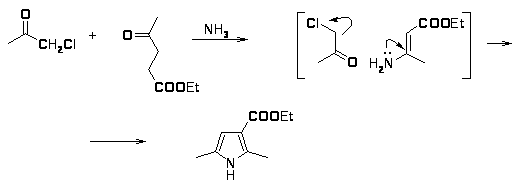
Было показано, что при конденсации 3-(-диэтиламино)этил-пентан-2,4-диона (3) изонитрозоацетоуксусным эфиром (1) наряду с 2,4 диметил 3 ( диэтиламино)этил-5 карбоэтокси-пирролом (8) образуется 3 ацетилпиррол (7) [17,18]. Если вместо ацетоуксусного эфира использовать малоновый эфир (2), то образуется 2,4-диметил-5-карбоэтоксипиррол (9).
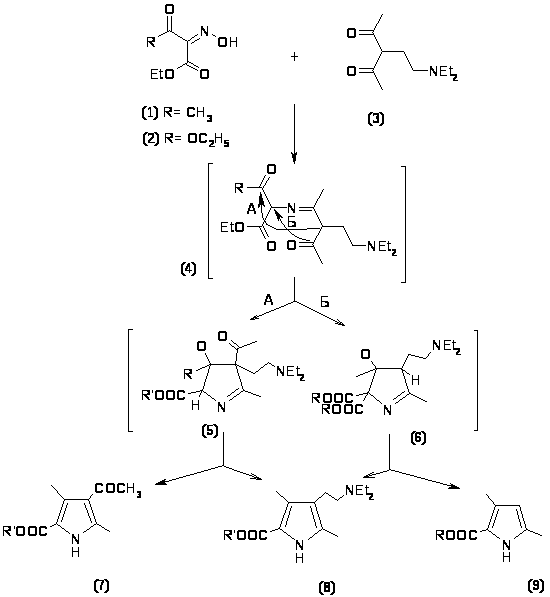
Реакция может протекать либо по пути А с участием ацильной группы; либо по пути Б, когда ацетильная группа участвует в построении гетероцикла. В случае циклизации пентандиона (3) с -диэтиламиноэтильной группой с изонитрозоацетоуксусным эфиром реакция осуществляется по механизму А. Конденсация пентандиона (3) с изинитрозодиэтилмалоновым эфиром происходит по пути Б.
Ещё одним классом соединений, широко используемых в синтезе пирролов, являются нитроалкены [19]. Так при конденсации -метил--нитростирена (10) с метилацетоацетатом (11) образуется промежуточное соединение (12). При конденсации этого же стирена с этилацетоацетатом (13) образуется другое промежуточное соединение (14), однако оба этих промежуточных соединения дают пирролы одного строения.
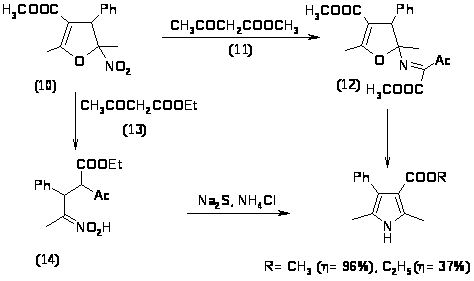
В последние годы, в связи с необходимостью введения в порфириновый макроцикл сложных ароматических систем, таких как фенантрен или фенантролин , нафталин и аценафталин, бензол [7,20,21], а также ацетали нитроацетальдегида [22] был разработан синтез пирролов, конденсированных по положениям с выше указанными ароматическими системами на основе метода Бартона Зарда, которые показали, что нитроалкены способны при конденсации с изоцианоацетатами в присутствии ненуклеофильного основания давать пиррол-2-карбоксилаты. Хотя сам нитробензол не реагирует таким образом, другие нитроарены с большим числом двойных связей конденсируются с изоцианоацетатами, давая соответствующие пирролы с хорошим выходом до 50%.
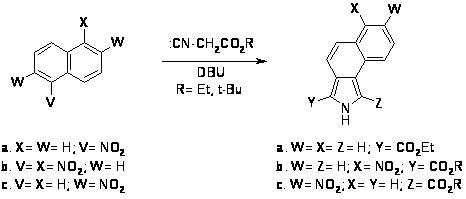
Также были предприняты попытки синтеза пирролов, конденсированных с гетероароматическими и гетероциклическими системами, однако нитротиофен, нитрофуран и нитропиридин дали в качестве продуктов смолу, хотя 4-нитро-2,1,3-бензотиадиазол при конденсации с изоцианоацетатом дает в качестве одного из продуктов соответствующий пиррол [21].
2.1.2. Конденсации, при которых в готовый углеродный скелет вводится атом азота при помощи аммиака или аминов.
Наиболее важной в этой группе реакций является взаимодействие 1,4-дикарбонильных соединений с аммиаком по Паалю-Кнорру [19,23]. Механизм реакции, очевидно, включает нуклеофильное присоединение аммиака к двум карбонильным атомам углерода и последующее отщепление двух молекул воды [24].
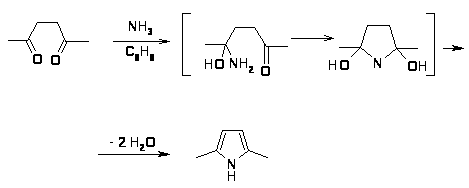
Отмечено также, что реакция может протекать с ацетатом аммония c хорошим выходом (~ 70%), причем, чем более электроноакцепторные заместители в 1,4-дикетоне, тем в более жестких условиях протекает реакция [25].
К этой же группе реакций можно отнести получение пирролов по Ганчу из галогенкетонов, -кетоэфиров и аммиака [26].
Согласно предложенному механизму, сначала происходит образование С-С связи и возникает -дикетон, который далее реагирует с амином. К реакциям этой группы относится также взаимодействие аммиака и аминов с полиокси- и полигалоидными соединениями [27].
1H-пирролы также образуются в результате термической 1,5-сигматропной перегруппировки 3Н-пирролов, которые образуются по реакции Пааля-Кнорра [28].
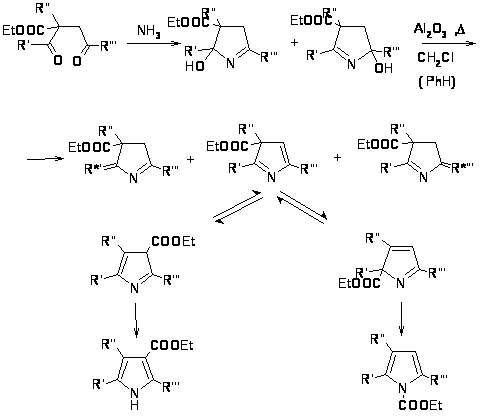
Однако представленный выше метод не отличается хорошей селективностью, в результате реакции образуются изомеры, которые дают множество побочных продуктов при дальнейшей обработке. Низкий выход целевого продукта (~ 20%), а также общее время протекания всех стадий (от 90 до 160 часов) делают этот метод малоприемлемым для широкого применения.
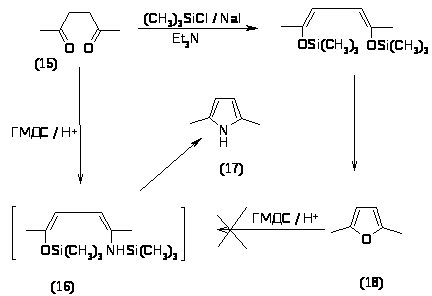
Метод получения 2,5-диметилпиррола (17) из 2,5-гександиона (15) и гексаметилдисилазана (ГМДС) в присутствии трифторметансульфокислоты является отличным по выходу-100%, но дорогим из-за использования трифторметансульфокислоты. Реакция протекает через промежуточное соединение (16). Попытки получить 2,5-диметилпиррол (17) из 2,5 диметилфурана (18) через это соединение закончились неудачно [29].
2.1.3. Реакции присоединения аминов по кратным связям.
При реакции эфиров ацетиленкарбоновой кислоты с различными нуклеофилами образуются производные пиррола [30]. Первая стадия заключается в присоединении по Михаэлю, образующееся при этом соединение циклизуется затем по ниже приведенному механизму.
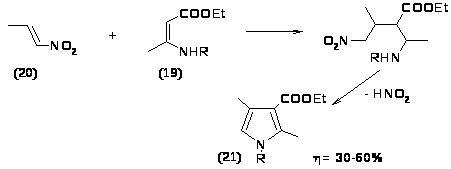
К этой же группе реакций можно отнести образование производных пиррола из N замещенных эфиров -аминокротоновой кислоты (19) и 1-нитропропена (20), которые в реакции типа Михаэля дают аддукт, превращающийся в пирролин, окисляющийся затем в пиррол (21) [31,32].
Производные пиррола из эфиров N-тозилглицина и ,-ненасыщенных кетонов являются промежуточными в синтезе порфиринов [33].
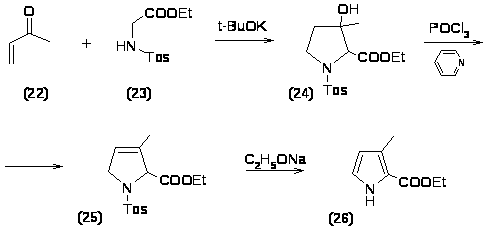
Присоединение по Михаэлю эфира N-тозилглицина (23) к кетону (22) в присутствии третбутилата калия с последующей внутримолекулярной альдольной конденсацией приводит к образованию гидроксипирролидина (24), при дегидрогалогенировании которого образуется Δ3-пирролин (25); отщепление от него п-толуолсульфиновой кислоты под действием этилата натрия приводит к пирролу (26) с удовлетворительным выходом ~ 30-40% [16].
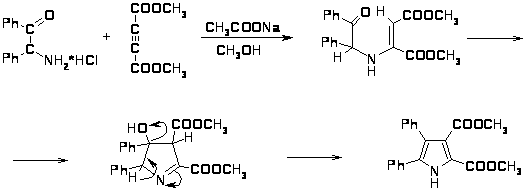
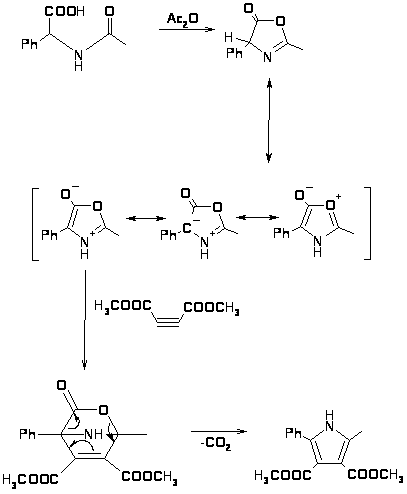
Ещё одним способом получения пирролов является получение пирролов по Хьюгенсу с помощью 1,3-диполярного присоединения диэфиров ацетилендикарбоновых кислот к азолактонам, образующимся в результате циклизации N-замещенных фенилглицинов в присутствии уксусного ангидрида [16]. В результате таутомерии азолактон находится в равновесии с ароматическим азометинилидом, к которому присоединяется диэфир ацетилендикарбоновой кислоты с образованием бициклического интермедиата, при отщеплении от которого CO2 образуется тетразамещенный пиррол с выходом 70-80% [34].
.
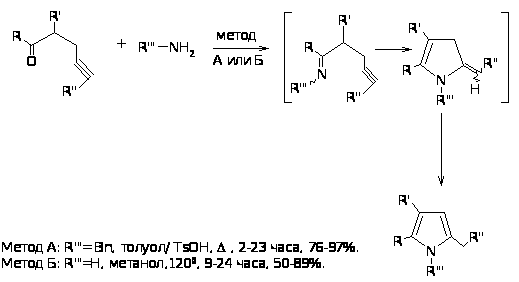
Синтез замещенных пирролов может быть также осуществлен с хорошим выходом реакцией γ-кетоалкинов с бензиламином или аммиаком. Механизм реакции, вероятно, включает образование имина, который подвергается 5-экзодициклизации с последующей изомеризацией давая пиррол [35].
В последние годы было обнаружено, что при взаимодействии 1,4-дикарбонильных соединений с солями аммония по Паалю-Кнорру, могут образовываться как производные пиррола, так и производные фурана [25, 36].
.
2.1.4. Реакции образования циклов в результате внутримолекулярной конденсации.
Одним из важных ограничений синтеза пирролов по Кнорру является склонность аминокетонов к автоконденсации. Поэтому был разработан метод синтеза пирролов из аминоамидов Вейнреба, которые проявляют низкую склонность к автоконденсации и возможность превращения в кетоны, которые затем циклизуются давая соответствующие пирролы.
Ключевой стадией процесса является превращение N метокси N метил енамино-карбоамидов (27) в карбонильные производные (28) под действием металлорганических соединений. На этой стадии металлорганическое соединение (МеLi, MeMgI) селективно атакует амидную группу. При этом сначала под действием металорганического соединения отщепляется протон, давая сопряженный анион (30) к которому присоединяется металлорганическое соединение, давая интермедиат (31) при гидролизе которого образуется карбонильное соединение (28), причем здесь же может происходить частичная или полная циклизация до пиррола (29). Дальнейшая циклизация карбонильного соединения (28) до пиррола (29) происходит в условиях щелочного катализа (NaOC2H5/ C2H5OH), выходы пирролов составляют 60-70% в зависимости от заместителей [37, 38].
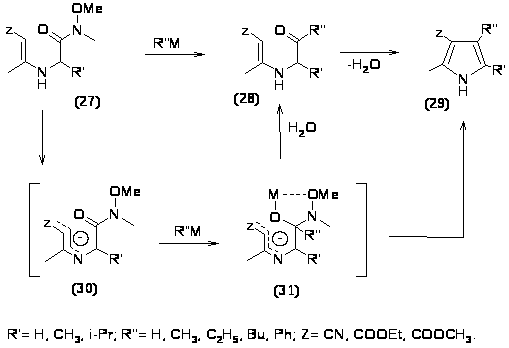
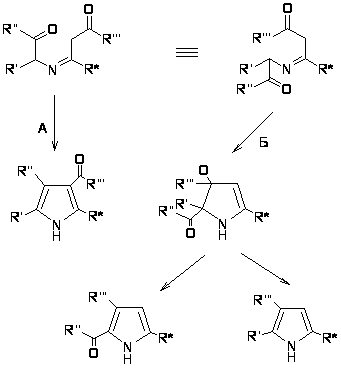
В данном случае из-за природы R1, R2 и Z происходит циклизация по Кнорру, однако если Z= ацил, то циклизация может происходить как по Кнорру (путь А), так и по Фишеру-Финку (путь Б) [18].
Ещё одним способом получения пирролов является йодоциклизация алкенилзамещенных -енаминоэфиров и кетонов с последующим дегидрогалогенированием. Основой для реакции циклизации является имин-енаминное таутомерное равновесие, в котором преобладает енамин.
Было показано, что продукт электрофильной циклизации зависит от относительного положения алкенильной цепи в енаминокетоне, так из -алкенил-аминокетонов образуется дигидропирролы (32), а из γ-алкенил--аминокетонов- 2-метиленпирролидины (33) [39].
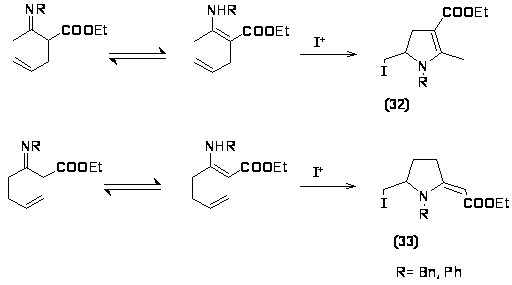
Также было отмечено, что скорость йодоциклизации зависит от заместителя у атома азота, в случае наличия сопряжения скорость увеличивается в 10-20 раз [40].
Дегидрогалогенирование дигидропиррола производят с помощью основания или без него, причем скорость дегидрогалогенирования подчиняется тем же закономерностям, что и скорость йодоциклизации.
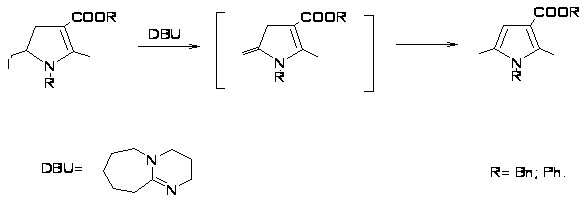
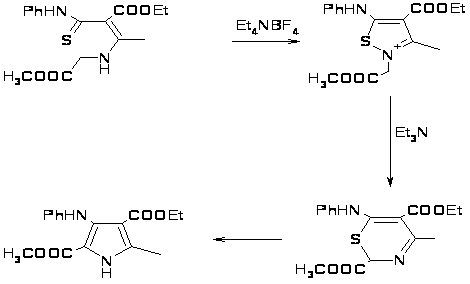
Широко используемым может быть метод получения тетразамещенных пирролов из ненасыщенных тиоамидов, включающий образование изотиазолевых солей в качестве интермедиатов в синтезе тиазинов, при удалении из которых серы образуются тетразамещенные пирролы [23].
Из- за сложностей возникающих при попытках получить аминопирролы из незамещенных пирролов, был разработан метод синтеза 3-аминопирролов с хорошими выходами при помощи циклизации Торпа Циглера. Метод основан на N-алкилировании -енаминонитрилов - галогенкетонами с последующей внутримолекулярной конденсацией по Торпу Циглеру [41,42].
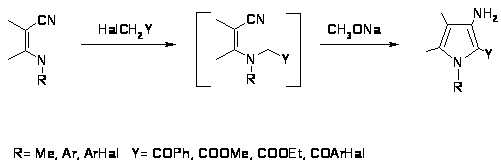
Основным требованием данной реакции является выбор алкилирующих агентов имеющих активное метиленовое звено, необходимое для протекания последующей внутримолекулярной конденсации, которая протекает спонтанно или в условиях щелочного катализа метилатом натрия.
2.2. Методы синтеза порфиринов.
Характерной чертой современной химии порфиринов является наличие значительного числа разнообразных методов построения порфиринового макроцикла. Наряду с методами, предложенными в последние годы, достаточно широко используются способы, разработанные в 20-30 годы. Новые методы, потеснив старые, полностью их не исключили. Такое, в общем, не характерное для иных классов биологически-активных соединений, состояние связано, главным образом, с тем, что синтез порфиринов зависит от характера имеющихся в порфирине заместителей и их расположения в макроцикле.
Методы, позволяющие получать сравнительно простые порфирины, совершенно не применимы к более сложным природным порфиринам. И напротив, общие методы, предложенные в последние годы, нецелесообразно применять для получения ряда порфиринов, включая и некоторые природные, поскольку в этом случае неоправданно усложняется синтез исходных пирролов [43].
Синтез порфиринов можно осуществлять двумя основными методами:
синтез функциональных порфиринов из непорфириновых предшественников;
синтез функциональных порфиринов из заранее полученных незамещенных порфиринов.
Сначала рассмотрим первый, наиболее распространенный, метод получения порфиринов.
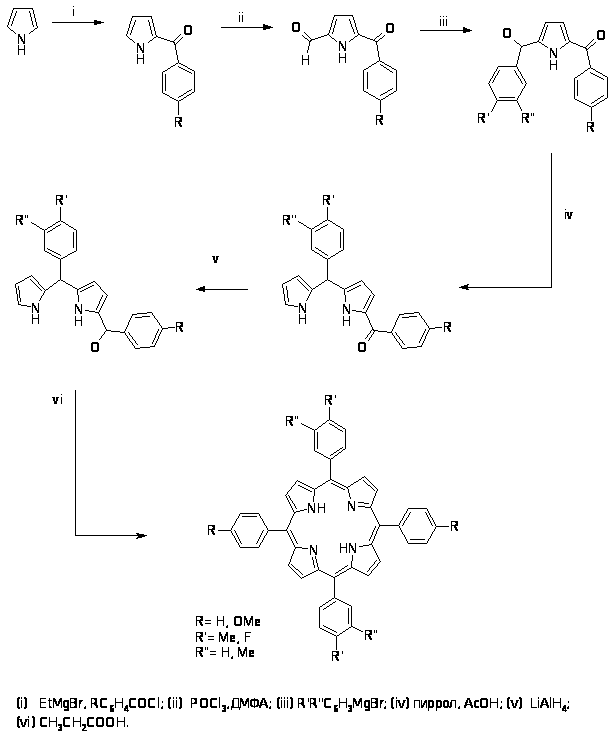
2.2.1. Метод Адлера-Лонго.
Метод Адлера-Лонго разрабатывался в середине ХХ века, его часто используют для получения несимметричных замещенных тетраарилпорфиринов. Главной особенностью метода является получение полностью мезо - замещенного порфирина, в мезо положение можно ввести как ароматические заместители [44,45], так и алифатические [46].
Суть метода заключается в конденсации α–незамещенного пиррола с альдегидом, при этом в конденсацию можно вводить смесь различных пирролов и альдегидов, но при этом образуется смесь из всех возможных комбинаций порфиринов и расширенных порфириновых циклов, содержащих 5 и более пиррольных фрагментов [47,48].
Монопиррольная тетрамеризация широко используется, когда целевым является высокосимметричный порфирин, например октаэтилпорфирин или тетрафенилпорфирин, при этом в конденсацию вводят пирролы, у которых в одном из -положений находится метиленовая группа с легко уходящим остатком, а другое – свободно [49,50].
В последние годы были разработаны методы получения несимметричных порфиринов данным способом, но они требуют тщательного хроматографического разделения продуктов реакции [51].
Тем не менее, оказалось возможным предсказать образование требуемого порфирина, если использовать пиррол, содержащий во втором положении такую уходящую группу, которая была бы настолько реакционноспособна, чтобы ее можно было заместить без кислотного катализа, а образующийся порфириноген быстро бы окислялся в порфирин. Идеальной уходящей группой оказалась диметиламинометильная группа, благодаря которой конденсацию можно проводить действительно в нейтральных условиях. Более того, оказалось возможным получить региохимически чистый порфирин (с одинаковыми пиррольными кольцами напротив друг друга) при использовании пиррола с двумя диметиламинометильными группами и пиррола, не содержащего заместителей в -положениях пиррольного цикла, выход составил 10-20% [52].
При необходимости реакционную способность диалкиламинометильной группы увеличивают путем проведения реакции кватернизации, при этом выход порфириновой конденсации увеличивается до 20%.
2.2.2. “2+2” порфириновый синтез.
Этот метод основан на конденсации двух молекул дипиррометанов. Существенным преимуществом синтезов порфиринов через дипирролилметаны является то, что последние получаются с достаточно высокими выходами, особенно при наличии электронодонорных заместителей в пиррольном цикле [53].
Первоначальный метод Макдональда включал использование одного дипиррометана, содержащего две формильные группы в α положениях и другого α свободного или содержащего карбоксильные группы дипиррометана [33,54,55].
Линдсей с соавторами синтезировали порфирины, содержащие 4 различных мезозаместителя, с помощью модифицированного метода Макдональда [56,57]. В качестве заместителей в мезо- положениях выступали алкил- и галоген замещенные бензолы, выход требуемого продукта достаточно высок для порфиринового синтеза и составил 14%.
Оно и сотрудники использовали 5,5’-бис(гидроксиметил)дипиррометаны для получения несимметричных порфириновых димеров [58]. Андерсон синтезировал порфириновые димеры, используя замещенные дипиррометан и триметилсилилпропиналь, как один из углеродных фрагментов. Несимметричные мезо-арилпорфирины с заместителями в β положениях пирольного цикла также были синтезированы данным способом. Огоши с соавторами синтезировали 5,15-дифенил-10-р-хлорфенил-2,3,17,18-тетраэтилпорфирин с выходом 9% [59].
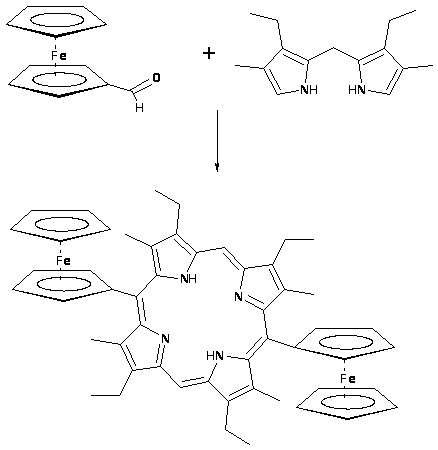
Другой модификацией этого метода является синтез Смита, заключающийся в конденсации α свободных дипиррометанов и ароматического альдегида [60,61]. Возможна конденсация двух молекул пиррометана, содержащего в одном из α положений карбонильную группу, при этом получаются зеркальносимметричные порфирины. В последние годы в качестве углеродного фрагмента, соединяющего две молекулы пиррометана, используют не только ароматические альдегиды, но и, например известны синтезы, в которых в конденсацию вводят карбальдегиды ферроцена, выход этих реакций достигает 50% [62]. Порфирины, полученные этим способом, например, мезо-тетракис(1,3-диметилимидазол-2-ил)порфирин и мезо-тетракис(1,2-диметилпиразол-4-ил)порфирин, способны различным путем встраиваться в молекулу ДНК и могут быть использованы в качестве молекулярного транспорта [63].
Смит и сотрудники синтезировали мезоарил-замещенные порфирины, состоящие из двух зеркально симметричных фрагментов. Авторами были получены тетраарилпорфирины с различными арильными заместителями по ниже приведенной схеме. 5,15-Ди(4-толил)-10,20-дифенилпорфирин получают данным способом с 31% выходом, а 5,15-бис(4-фторо-3-метилфенил)-10,20-(4-метоксифенил)порфирин – с выходом 24% [60,64].
В дипирролилметане (34), имеющем две близкие по реакционной способности группы в положениях 5 и 5’, проводят активацию формильной группы, превращая ее через основание Шиффа (35) в тиоальдегид (36). Конденсация соединений (36) и (37), проводимая в мягких условиях, приводит лишь к одному соединению (38), замыкание и перегруппировка последнего приводят к хлорину (39) с высоким выходом.
2.2.2.1 Синтез порфиринов через b-билены.
В настоящее время известно значительное число методов получения как частично симметричных, так и полностью несимметричных порфиринов через b-билены.
Этот метод достаточно широко используется для синтеза сложных порфиринов, этому способствует ряд обстоятельств: а) более легкий синтез исходных дипирролилметанов по сравнению с получением аналогичных 5,5’-диалкоксикарбонильных соединений, которые необходимы в методе Макдональда; б) отсутствие гексапирродиенов при синтезе биленов; в) возможность получения полностью несимметричных порфиринов. Следует отметить, что при синтезе диацетилпорфирина было показано, что при получении порфиринов с одной и особенно с двумя электроотрицательными группами синтез биленов надо планировать таким образом, чтобы эти заместители находились в крайних пиррольных кольцах. При наличии подобных заместителей в дипирролилметановом фрагменте билена повышалась чувствительность метинового мостика к электрофильным реакциям. Возможность побочных реакций особенно возрастала, когда два таких заместителя расположены в противоположных пиррольных кольцах. С учетом сказанного был синтезирован замещенный цитопорфирин (40), который является ключевым соединением при получении порфирина α, выход составил 30%.
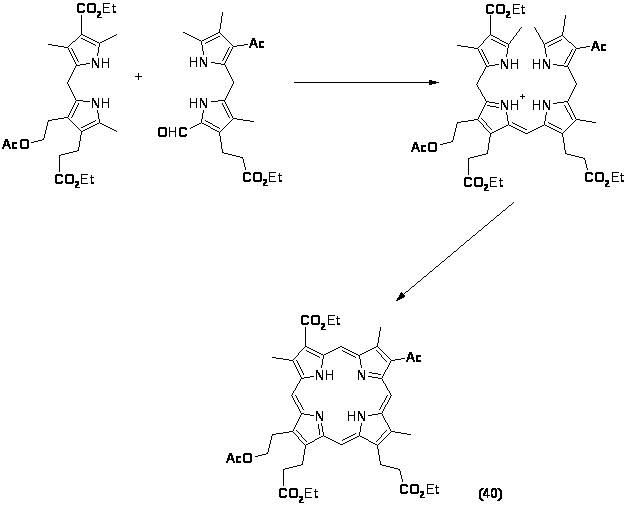
В заключение можно отметить, что использование b биленов в качестве промежуточных соединений позволяет получать широкий круг порфиринов с различными заместителями. Среди недостатков этого метода следует отметить недостаточную устойчивость b-биленов и их подверженность нежелательным превращениям.
2.2.2.2. Циклизация а,с-биладиенов.
Биладиены, среди прочих тетрапиррольных структур, играют важную роль в природе (желчные пигменты и протеины) и являются интермедиатами в синтетических подходах к природным порфиринам [65]. В 1966 году Джонсон предложил оригинальный метод синтеза порфиринов, который позволяет получать как симметричные, так и несимметричные порфирины [66].
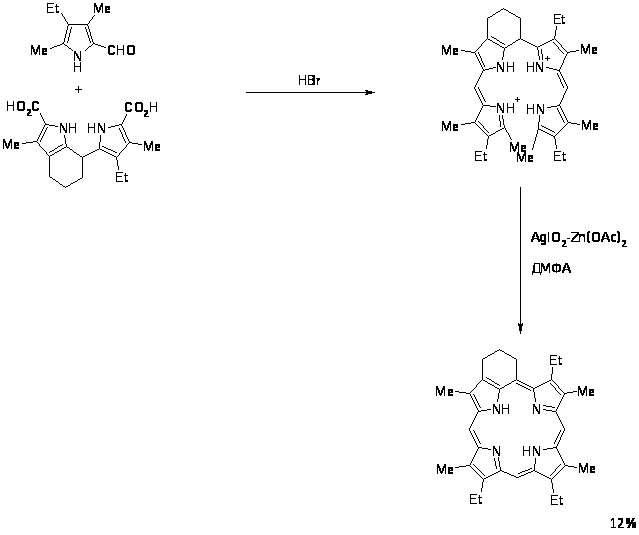
Данный способ нашел применение для алкилзамещенных порфиринов. Выход выше изображенного порфирина на последней стадии составил ~ 70%. На основе а,с- биладиенов также были синтезированы производные мезо-аминопорфирина при аномальной циклизации 1,19-диметил-а,с-диметил-а,с-биладиенов с Cu(OAc)2 в ДМФА, причем мезо-углеродный атом порфиринового макроцикла образуется из карбонильного углерода ДМФА [8,67,68].
2.2.3. “3+1” порфириновый синтез.
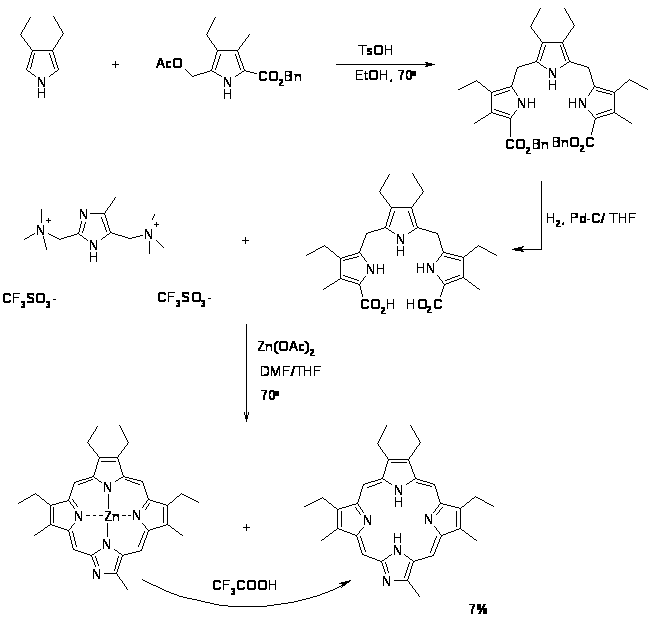
Этот способ позволяет получать как симметричные, так и несимметричные порфирины и их аналоги, причем, выход может достигать отличных для порфиринового синтеза значений 40-50% [69]. Метод состоит в конденсации трипирана с 2,5 диформилпирролом [70] или 2,5-бис[(N,N-диметиламино)метил]пирролом [52,71]. Синтез с 2,5 диформилпирролом используется наиболее часто, его проводят, когда исходные субстраты способны выдержать условия кислотного катализа, либо при однозначном образовании одного изомера порфирина или возможности легкого разделения смеси изомеров. Недостатком выше описанного метода, являлась сложность синтеза 2,5 диформилпирролов, однако в последние годы был разработан достаточно простой и надежный способ позволяющий получать пиррол дикарбальдегиды с хорошим выходом [6,70,72,73].
Другим возможным вариантом «3+1» порфиринового синтеза является использование 2,5 бис[(N,N диметиламино)метил]пиррола, при этом реакция проводится в более мягких условиях, за счет чего достигается образование одного изомера порфирина, а не всех возможных как при использовании 2,5 диформилпирролов [52]. При помощи этого метода стало возможным синтезировать азапорфирины, содержащие в качестве одного из пиррольных циклов имидазольный фрагмент, с удовлетворительным выходом [71].
3. Обсуждение результатов.
Порфириновые макроциклы сопряженные по -положениям пиррольного кольца с ароматическими и гетероциклическими системами нашли применение в качестве молекулярных зондов, высокоэффективных катализаторов, фотосенсоров в фотодинамической терапии рака и красителей поглощающих в ближнем ИК диапазоне спектра. Целью данной работы является поиск подходов к синтезу сопряженной системы пиррола с гетероциклическим фрагментом для последующего введения ее в порфириновый цикл.
Из литературных источников известно, что существует несколько подходов к синтезу порфиринового макроцикла, наиболее часто используемыми являются методы “2+2” и “3+1. Последний метод был выбран как наиболее подходящий. Для его реализации необходимо было синтезировать -алкил или -незамещенный трипиран и пиррол, содержащий в -положениях функциональные заместители. С этой целью был синтезирован трипиран по следующей схеме:
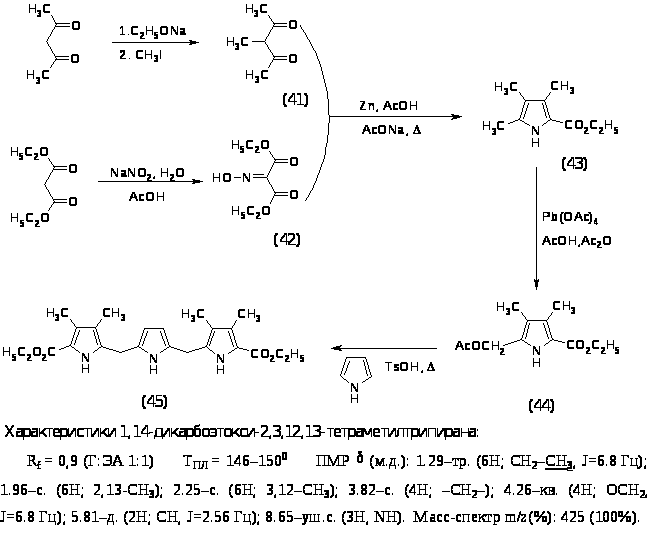
Алкилирование ацетилацетона проводили по стандартной методике - с применением на первой стадии этилата натрия и последующей обработкой метилиодидом при 400 в течение 1 часа, выход 3–метил–2,4–пентандиона (41) составил 48%. Синтез 2 карбоэтокси-3,4,5 триметилпиррола (43) проводили из 3-метил-2,4-пентандиона (41) и изонитрозомалонового эфира (42) конденсацией в уксусной кислоте в присутствии цинка и ацетата натрия при нагревании до 900 в течение 1 часа, выход продукта составил 71%. 2 ацетоксиметил–3,4–диметил–5–карбоэтоксипиррол (44) получали окислением 2 карбоэтокси-3,4,5-триметилпиррола (43) тетраацетатом свинца в смеси уксусной кислоты и уксусного ангидрида при комнатной температуре в течение 2 часов, выход 2 ацетоксиметил–3,4–диметил–5–карбоэтоксипиррола (44) составил 82%. Синтез 1,14-дикарбоэтокси-2,3,12,13-тетраметил трипирана (45) проводили конденсацией в метаноле 1 эквивалента пиррола и 2 эквивалентов 2 ацетоксиметил–3,4–диметил–5–карбоэтоксипиррола (44) в присутствии толуолсульфокислоты при нагревании до 600 в течение 7 часов, выход 1,14-дикарбоэтокси-2,3,12,13-тетраметил трипирана (45) составил 51%. Все синтезированные продукты были охарактеризованы спектральными методами и были определены их физико-химические константы.
Следующим этапом работы была разработка методов получения монопиррольных интермедиатов для получения сопряженной системы, включающей два гетероциклических фрагмента. Из литературных данных известно, что существует 2 подхода к синтезу сопряженной системы пиррола с гетероциклическим фрагментом:
К готовому гетероциклическому фрагменту, используя реакцию Бартона-Зарда, присоединяют пиррольный цикл.
К готовому пиррольному циклу, имеющему функциональные группы в -положениях наращивают гетероциклический фрагмент.
На основе первого подхода было решено провести конденсацию 2-метил-6-нитробензотиазола (47) и этилового эфира изоциануксусной кислоты (51) в присутствии сильных ненуклеофильных оснований, т.е. в условиях реакции Бартона-Зарда [7,20,21].
Нитрование 2-метилбензотиазола (46) проводили нитрующей смесью при нагревании до 900 в течение 5 часов, выход 2-метил-6-нитробензотиазола (47) составил 20%.
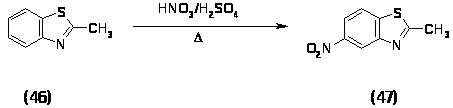
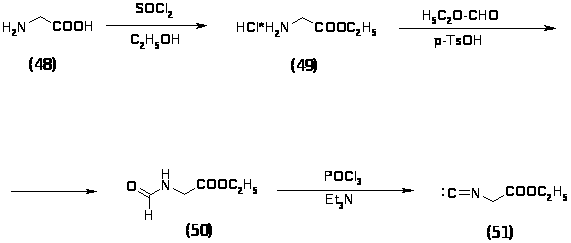
Исходным соединением в синтезе этилового эфира изоциануксусной кислоты (51) являлся глицин (48), который превращали в хлоргидрат глицинэтилового эфира (49) действием тионилхлорида в этаноле при кипячении с обратным холодильником в течение 2 часов. Выход продукта составил 94%. Полученный хлоргидрат глицинэтилового эфира (49) кипятили в этилортоформиате в присутствии толуолсульфокислоты и триэтиламина в течение 20 часов. Получили этиловый эфир N-формилглицина (50) с выходом 66%, который после обработки POCl3 в триэтиламине и дал этиловый эфир изоциануксусной кислоты (51) с выходом 76%. Все синтезированные продукты были охарактеризованы спектральными методами и были определены их физико-химические константы [16].
Полученные 2-метил-6-нитробензотиазол (47) и этиловый эфир изоциануксусной кислоты (51) растворяли в абсолютном ТГФ и вводили в конденсацию в присутствии сильных оснований (условия проведения реакций и обработка приведены в таблице). Однако реакция протекала плохо, в основном возвращался исходный 2-метил-6-нитробензотиазол (47) и получалось множество продуктов, суммарный вес которых незначителен.
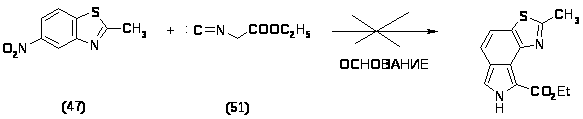
Реакция 2-метил-6-нитробензотиазола и этилового эфира изоциануксусной кислоты.
|
Отношение 2 метил 6 нитробензотиазола к этиловому эфиру изоциануксусной кислоты. |
Растворитель, объем (на 0,1 г 2-метил-6-нитробензотиазола) (мл) |
Основание, отношение к 2 метил 6 нитробензотиазолу |
Условия проведения реакции, обработка. |
|
1:1.1 |
ТГФ абс. , 50 мл |
DBU, 1: 1.1 |
100 часов при комнатной температуре. |
|
1:1.1 |
ТГФ абс. , 50 мл |
DBU, 1: 1.1 |
Кипячение 12 часов. |
|
1:1.1 |
ТГФ абс. , 50 мл |
(Et2N)3P=NEt, 1:1 |
24 часа при 200 и 4 часа при 600, разбавляют CHCl3 и промывают водой. |
|
1:1.1 |
ТГФ абс. , 50 мл |
(Et2N)3P=NEt, 1:2 |
200 часов при 200, разбавляют CHCl3 и промывают водой. |
|
1:1.1 |
ТГФ абс. , 50 мл |
(Et2N)3P=NEt, 1:2 |
200 часов при 500, разбавляют CHCl3 и промывают водой. |
|
1:1.1 |
ТГФ абс. , 40 мл |
NaH, 1:1 |
72 часа при при 200, разбавляют CHCl3 и промывают водой. |
|
1:1.1 |
ТГФ абс. , 40 мл |
NaH, 1:2 |
72 часа при при 200, разбавляют CHCl3 и промывают водой. |
|
1:1.1 |
ТГФ абс. , 40 мл |
NaH, 1:2 |
72 часа при при 500, разбавляют CHCl3 и промывают водой. |
Использование данного подхода было признано не эффективным, таким образом, было решено перейти к второму подходу. Предполагалось присоединить тиазольный или имидазольный фрагмент по -положениям пиррола. С этой целью необходимо было разработать удобные методы синтеза ,’-диаминопиррола или -галоген-’-аминопиррола. Аминогруппы предполагалось получать путем восстановления нитрогрупп, и задача сводилась к получению нитропирролов.
Первоначально для нитрования был выбран 2,5-диметилпиррол (56), который был синтезирован по следующей схеме:
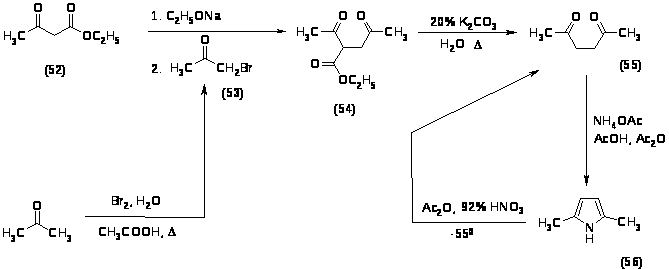
Алкилирование ацетоуксусного эфира (52) проводили бромацетоном (53), полученным предварительно с выходом 40% при обработке ацетона бромом в водной уксусной кислоте при 700 в течение 2 часов. Алкилирование ацетоуксусного эфира (52) проводили двумя способами: 1) с использованием катализатора межфазного переноса [74]; 2) по стандартной методике с использованием этилата натрия. Выход реакции алкилирования по первому методу составил 20%, а по второму – 53%, поэтому для наработки ацетонилацетоуксусного эфира (54) был выбран последний способ. Полученный ацетонилацетоуксусный эфир (54) декарбоксилировали при кипячении с обратным холодильником в течение 1 часа 20% водным раствором поташа. Выход 2,5-гександиона (55) составил 65%. Замыкание полученного 2,5-гександиона (55) в пиррольный цикл проводили ацетатом аммония в смеси уксусной кислоты и уксусного ангидрида при комнатной температуре в течение 0,5 часа. Выход 2,5-диметилпиррола (56) составил 57%. Нитрование 2,5-диметилпиррола (56) проводили при –550 смесью 92% HNO3 и уксусного ангидрида, однако, даже в таких мягких условиях произошло раскрытие цикла, и в качестве продукта был выделен 2,5-гександион (55). Все синтезированные продукты были охарактеризованы спектральными методами и были определены их физико-химические константы.
Так как известно, что введение акцепторных заместителей приводит к стабилизации пиррольного цикла, поэтому было решено вводить нитрогруппы в пиррол, содержащий акцепторные группы. С этой целью в качестве соединения для введения нитрогрупп был выбран 2,5-диформилпиррол (61).
Существующие методики получения 2,5-диформилпирролов [75-82] отличаются многостадийностью, малой доступностью исходных реагентов и умеренными выходами. Поэтому необходимо было разработать простой и эффективный метод получения этого соединения.
В качестве исходного соединения был выбран пиррол. Введение первой формильной группы не представляет труда и описано в литературе [83]. Формилирование пиррола (57) проводили по стандартной методике комплексом Вильсмеера при 350 в течение 0,5 часа, гидролиз проводили при кипячении в водном растворе ацетата натрия в течение 0,5 часа, выход 2-формилпиррола (58) составил 64%. Прямое формилирование 2-формилпиррола (58) приводит главным образом к 2,4-диформилпирролу и к 0,3% 2,5-диформилпиррола (61) [75]. Для введения альдегидной группы в 5-положение необходимо было ввести группу с одной стороны направляющую в это положение, а с другой стороны легко снимаемую в результате обработки. В качестве такой группы была выбрана дикарбоэтоксивинильная группа. 2-(Пиррол-2-илметилен)малоноат (59) получали при кипячении в бензоле 2-формилпиррола (58) и диэтилмалонового эфира в присутствии пиперидина и уксусной кислоты в течение 1 часа. Продукт без дополнительной очистки был направлен на следующую стадию.
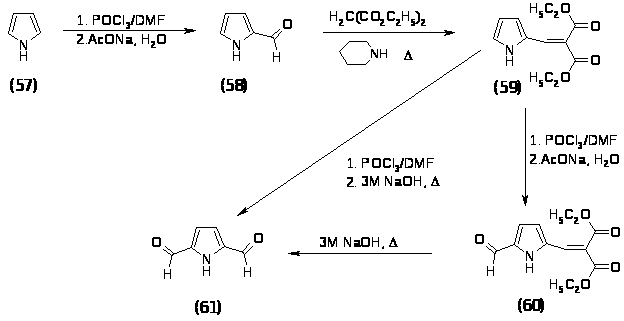
Формилирование 2-(пиррол-2-илметилен)малоноата (59) проводили комплексом Вильсмеера при 400. В зависимости от условий проведения гидролиза полученной соли возможно выделить продукт формилирования без снятия защитной группы. Так при проведении гидролиза насыщенным раствором ацетата натрия при комнатной температуре образуется 2 [(5 формилпиррол-2-ил)метилен]малоноат (60) с выходом 55%, который был полностью охарактеризован спектральными методами и физико-химическими методами и имеет следующие характеристики: Rf = 0,7 (Хл:МеОН 9:1); ТПЛ = 98–1000; ПМР (м.д.): 1.31–тр. (3H; CH3CH3, J=7.26 Гц); 1.37–тр. (3H; CH3CH3, J=7.26 Гц); 4.30–кв. (2H; CH3, J=7.26 Гц); 4.38–кв. (2H; CH3, J=7.26 Гц); 6.68–д.д. (1H; CH, J=2.14 Гц); 6.94–д.д. (1H; CH, J=2.14 Гц); 7.57–c. (-CH=C); 9.65 с. (CHO); 11.53 уш.c. (1H; NH); 13С ЯМР (м.д.): 13.88 (OCH3CH3); 13.99 (OCH3CH3’); 61.60 (О CH3); 62.11 (О CH3’); 119.53 (=С-); 120.49 (С 4); 121.92 (С-3); 131.85 (C-5); 134.09 (-C=); 135.63 (C-2); 163.30 (С=О); 166.66 (С=О’); 179.78 (CHO). ИК (вазелиновое масло) (cм 1): 3300, 1730, 1700, 1670, 1620, 1550. Масс-спектр m/z (%): 265 (71%), 173 (100%), 145 (46%), 119 (30%), 91 (28%), 65 (30%), 39(15%).
При гидролизе с применением 3М NaOH и кипячении в течение 1 часа образуется 2,5-диформилпиррол (61) с выходом 53%, который был охарактеризован спектральными методами и физико-химическими методами и имеет следующие характеристики: Rf = 0,3 (Хл:МеОН 9:1); ТПЛ = 112–1140; ПМР (м.д.): 7.02–д. (2H; CH, J=2 Гц); 9.77 с. (2H; CHO); 10.38 уш.c. (1H; NH); ИК (вазелиновое масло) (cм 1): 3140, 1720, 1700. Масс-спектр m/z (%): 123 (65%), 94 (18%), 66 (60%), 39(100%).
Полученный 2,5-диформилпиррол (61) предполагалось пронитровать в -положение. В качестве нитрующего агента использовали смесь 98% HNO3 и уксусного ангидрида. По литературным данным известно, что пиррольный цикл, содержащий акцепторные группы должен быть устойчив в указанных условиях. Действительно разрушения цикла не происходило, однако одна из альдегидных групп окислилась, о чем свидетельствует спектр ПМР выделенного продукта (62): ПМР (CDCl3-CD3OD) (м.д.): 6.15–д. (1H; CH, J=4 Гц); 6.68–д. (1H; CH, J=4 Гц); 7.94 с. (1H; CHO).
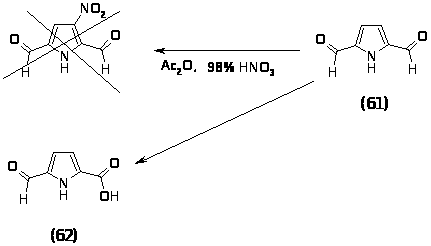
Поэтому было решено проводить реакцию нитрования 2,5-диметил-3-карбоэтоксипиррола (63). Данный пиррол был получен конденсацией ацетонилацетоуксусного эфира (54) ацетатом аммония в смеси уксусной кислоты и уксусного ангидрида при комнатной температуре в течение 20 минут с выходом 76%, характеристики полученного продукта совпали с литературными данными [84].
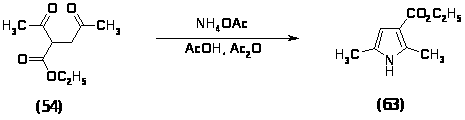
На этот раз прямое нитрование решили не использовать, так как велика вероятность раскрытия цикла и нитрогруппу решили вводить реакцией замещения галогена в -положении на нитрогруппу при помощи нитрита серебра. Реакцию проводили двумя способами:
одностадийный метод с использованием нитрита серебра и йода в ацетонитриле;
двух стадийный метод, включающий первоначальное введение галогена, который затем замещается на нитрогруппу.
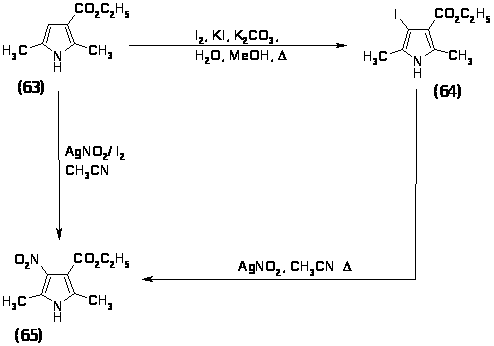
По первому способу введение нитрогруппы проводили обработкой 2,5-диметил-3-карбоэтоксипиррола (63) в ацетонитриле смесью нитрита серебра и йода в соотношении 2:1 при комнатной температуре в инертной атмосфере в течение 48 часов, выход 2,5-диметил-3-карбоэтокси-4-нитропиррола (65) составил 18%, соединение было полностью охарактеризовано спектральными методами и физико-химическими методами и имеет следующие характеристики: Rf=0.5(ПЭ:ЭА 1:1); Тпл= 110-1120 ; ПМР(CDCl3) (м.д.): 1.35 м.д.-тр.(3Н, СН2 СН3, J=7.1Гц); 2.35 м.д.-с.(3Н, СН3 ); 2.48 м.д.-с.(3Н, СН3); 4.32 м.д.-кв.(2Н, СН2-СН3, J=7.1Гц); 8.91 м.д.-уш.с.(1Н, NH). ИК(вазелиновое масло) (см–1) : 3300, 1720, 1680, 1600. Масс-спектр m/z(%): 212 (19%), 166 (40%), 122 (43%), 92 (45%), 66 (28%), 54 (19%), 42 (100%).
По второму способу сначала необходимо было получить -галогензамещенный пиррол. Иодирование 2,5-диметил-3-карбоэтоксипиррола (63) проводили в водно-метанольной смеси йодом в присутствии KI и поташа при 650 в течение 1,5 часов, выход 2,5-диметил-3-йод-4-карбоэтоксипиррола (64) составил 80%, соединение было полностью охарактеризовано спектральными методами и физико-химическими методами и имеет следующие характеристики: Rf = 0,7 (Г:ЭА 1:1); ТПЛ =116-1200; ПМР (CDCl3) (м.д.):1.34-тр. (3H; -CH3CH3; J=7.24 Гц) 2.22–с. (3H; CH3); 2.47–c. (3H; CH3); 4.26–кв. (2H; CH3; J=7.24 ГцРРHHsss); 8.18 уш.c. (1H; NH). 13С ЯМР (CDCl3) (м.д.): 13.89 (2-CH3); 14.19 (3-CH3); 14.30 (OCH3CH3); 59.58 (C-I); 63.03 ( CH3-); 129.48 (C-5); 129.55 (C-4); 135.58 (C-2); 164.62 (C=O). ИК(вазелиновое масло) (см 1): 3256, 1675, 1217, 1099, 1029, 773. Масс спектр m/z (%): 293 (40%), 279 (20%), 264 (42%), 248 (28%), 219 (8%), 127 (12%),122 (25%), 93 (30%), 67 (35%), 51 (55%), 42 (100%).
Реакцию замещения йода на нитрогруппу проводили при кипячении 2,5-диметил-3-йод-4-карбоэтоксипиррола (64) с нитритом серебра в ацетонитриле в течение 2 часов, выход 2,5-диметил-3-карбоэтокси-4-нитропиррола (65) составил 47%. Таким образом, второй метод оказался более предпочтительным, так как суммарный выход по этому способу составляет 37%, а по первому методу - 18%.
Также предполагалось омылить и декарбоксилировать полученный 2,5-диметил-3-йод-4-карбоэтоксипиррол (64) для дальнейшего введения нитрогруппы, однако омыление в различных условиях приводило лишь к отщеплению йода (условия проведения реакций и обработка приведены в таблице).
Омыление 2,5-диметил-3-иод-4-карбоэтоксипиррола.
|
Омыляющий реагент. |
V, (мл) |
Растворитель |
V (на 0,5 г пиррола, мл) |
Условия реакции, обработка. |
Время реакции. |
Продукт реакции омыления. |
|
H3PO4 |
2 мл |
– |
– |
1200 |
30 мин. |
Выделяется I2, пиррол осмоляется. |
|
20% раствор NaOH |
2.16 мл |
MeOH |
4 мл |
Кипячение, подкисляют конц. HCl |
5,5 ч. |
– “ – “ – |
|
20% раствор NaOH |
2.16 мл |
MeOH |
4 мл |
Кипячение, подкисляют H3PO4: H3O 1:5 |
4 ч. |
– “ – “ – |
|
H3SO4 конц. |
1.5 мл |
– |
– |
400, промывают водой |
30 мин. |
– “ – “ – |
|
30% раствор NaOH |
2.16 мл |
MeOH |
5 мл |
Кипячение, подкисляют конц. HCl |
2 ч. |
– “ – “ – |
|
H3SO4 конц. |
1.5 мл |
– |
– |
200, промывают водой |
2 ч. |
– “ – “ – |
Для синтеза нитро-галогензамещенного пиррола было решено использовать метод, описанный выше, поэтому необходимо было синтезировать ,’-дийодпиррол.
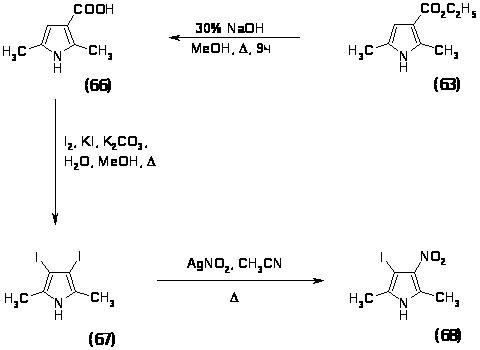
Указанное соединение получали в две стадии: первоначально проводили омыление 2,5-диметил-3-карбоэтоксипиррола (63) кипячением с 30% NaOH в метаноле в течение 9 часов, выход 2,5-диметил-3-карбоксипиррола (66) составил 65%; иодирование проводили в водно-метанольной смеси йодом в присутствии KI и поташа при 650 в течение 1,5 часов, выход 2,5-диметил-3,4-дийодпиррола (67) составил 45%. Соединение было охарактеризовано спектральными методами и физико-химическими методами и имеет следующие характеристики: Rf = 0,8 (Г:ЭА 1:3); ТПЛ =116-1200 (разл.); ПМР (м.д.): 2.28–с. (6H; CH3); 8.01 уш.c. (1H; NH); Масс спектр m/z (%): 347 (100%).
Реакцию замещения йода на нитрогруппу проводили по отработанной методике - при обработке 2,5-диметил-3,4-дийодпиррола (67) с нитритом серебра в ацетонитриле в течение 24 часов при комнатной температуре. В результате хроматографирования на колонке с силикагелем, было выделено 3 продукта (А, Б, В), которые были проанализированы с помощью Н1 ЯМР спектроскопии. ПМР (CD3OD) (м.д.): (А) 2.21-с.(1Н); 2.54-c. (1H). (Б)- 1.45-c.(4H); 1.47-c.(18H); 2.18-c.(6H); 2.41-c.(4H); 3.35-c.(4H). (B)-1.43-c.(9H); 1.46-д.(2H); 1.62-c.(1H); 1.87 c.(1H); 1.98-д.(2H); 2.42-c.(3H); 3.34-c.(3H); 6.51-c.(3H).
Основным является продукт (А) в ПМР-спектре которого наблюдалось 2 синглета одинаковой интенсивности в области 2.21 и 2.54 м.д., что позволило предположить о наличие двух неэквивалентных метильных группы в пиррольном кольце. Полученное соединение было проанализировано с помощью масс-спектрометрии MALDI. . Масс спектр m/z (%): 404 (100%); 388 (70%); 246 (42%). Полученные значения для молекулярных ионов оказались неожиданно высокими, что позволило предположить о наличии сложных структур, включающих в себя Ag+, что в последующем было подтверждено качественной реакцией на ионы серебра. Однако для установления структуры полученных комплексных соединений требуются дополнительные исследования.
4. Охрана труда
Введение
Работа на химических производствах связана с использованием агрессивных жидкостей и газов, высоких температур и других опасных и вредных факторов, влияющих на организм человека, поэтому необходимо уделять большое внимание вопросам охраны труда и защиты окружающей среды. Под охраной труда понимают систему мероприятий, обеспечивающих безопасность, сохранение здоровья и работоспособность человека в процессе труда. Охрана труда является неотъемлемой частью производственной деятельности и должна обеспечиваться на всех стадиях технологического процесса. Особенно важно учесть все вредные факторы при работе химика-технолога, труд которого неизбежно связан с токсическими и пожароопасными веществами. В ходе разработки методов синтеза и анализа новых препаратов, создания новых технологий должны быть тщательно продуманы все меры предосторожности, позволяющие исключить воздействие на человека опасных и вредных производственных факторов, обеспечить снижение травматизма и профессиональных заболеваний, обеспечить охрану окружающей среды.
Проведение любой научно-исследовательской работы в химической лаборатории неразрывно связано с приобретением необходимых навыков безопасной работы, изучением и последовательным соблюдением норм и правил техники безопасности, заботой об улучшении и оздоровлении условий труда, что очень актуально в настоящее время.
Рассматриваемая магистерская диссертация, посвященная синтезу пиррольных интермедиатов для высоко сопряженных порфиринов, выполнена на кафедре ХТТОС МГАТХТ им. М.В.Ломоносова. Ниже анализируются токсичные и пожароопасные свойства веществ, использованных в работе, а также условия, при которых проводился эксперимент и необходимые меры по охране труда.
Токсические и пожароопасные свойства веществ.
Под токсичностью химических веществ подразумевают их способность вызывать нарушения нормальной жизнедеятельности, приводить к патологическим изменениям в организме или вызывать гибель живого организма. Для характеристики вредности вещества применяется система ПДК в воздухе рабочей зоны. Пожароопасность определяется тем, что в работе используются легковоспламеняющиеся и горючие жидкости. В настоящей работе использовались пожароопасные и токсичные вещества. Все работы с ними проводились в вытяжном шкафу при полном отсутствии огня.
Токсикологические характеристики, величины ПДК и пожароопасные свойства веществ, используемых в работе, приведены в таблицах 1,2.
Таблица 1. Токсические свойства веществ [85,86].
|
Наименование вещества |
Характер воздействия на организм |
Меры и средства первой помощи |
ПДК, мг/м3 |
Класс опасности |
|
1 |
2 |
3 |
4 |
5 |
|
Азотная кислота |
Вызывает ожоги кожи, при вдыхании паров - удушье и кашель, при попадании внутрь - раздражение слизистых оболочек. |
При ожогах кожи - промыть большим количеством воды. |
5 |
III |
|
Ацетон |
Поражает нервную систему, наркотическое воздействие, оказывает влияние на функции почек. |
При инголяционном отравлении - промывание глаз водой, ингаляция кислородом; при перорильном - промывание желудка. |
200 |
IV |
|
Ацетонитрил |
Головная боль, апатия, тошнота. |
Вдыхание амилнитрита. |
10 |
Ш |
|
Бензол Толуол |
Действует на нервную систему, наркотик, снижает артериальное давление, нарушает дыхание, вызывает судороги, кровотечения в полости рта, влияет на состав крови, раздражает кожу. |
Искуственное дыхание, при попадании в органы пищеварения вызвать рвоту и дать слабительное. |
20 |
II |
|
1 |
2 |
3 |
4 |
5 |
|
Гексан Гептан |
Слабое воздействие на кожу, может вызвать временное опьянение. |
Свежий воздух. |
250 350 |
IV IV |
|
Диметилсульфоксид |
Малотоксичен. |
При попадании на кожу промыть водой. |
20 |
IV |
|
Диэтиловый эфир |
Наркотическое воздействие, слезотечение, возможно развитие токсического отека легких. |
Свежий воздух. При попадании внутрь - промывание желудка, рвота. |
300 |
IV |
|
Изопропиловый спирт |
Наркотик, влияет на зрение и центральную нервную систему. |
Свежий воздух, горячее молоко с содой. |
100 |
III |
|
Метанол |
Сильный нервный яд. При попадании внутрь вызывает потерю зрения, паралич дыхательных центров. |
Промывание желудка водой, этанолом, высокое положение головы, лед на голову. Вдыхание кислорода с 5% углекислоты, искуственное дыхание. |
5 |
III |
|
Натрия гидроксид Калия гидроксид |
Ожоги кожи и слизистых оболочек. |
Промыть струей воды, обработать борной кислотой. |
0,5 0,5 |
II II |
|
Пиперидин |
В больших дозах угнетает; вызывает отчетливую сосудистую реакцию с нарушением кровяного давления. |
При остром раздражении слизистых оболочек глаз промыть 2% раствором соды или борной кислоты. В глаза закапать 0,5% раствор дикаина. |
0,2 |
II |
|
1 |
2 |
3 |
4 |
5 |
|
SiO2 |
Общетоксическое действие, нарушение дыхательной функции легких (силикоз), раздражение легочной ткани острыми твердыми гранями частиц пыли. |
Устранение пылеобразования, использование индивидуальных защитных средств (очки, респиратор). |
1 |
II |
|
Соляная кислота |
Вызывает ожоги кожи, при вдыхании паров - удушье и кашель, при попадании внутрь - раздражение слизистых оболочек. |
При ожогах кожи - промыть большим количеством воды, затем 5% раствором бикарбоната натрия; свежий воздух. |
5 |
III |
|
Уксусная кислота |
Сильное раздражающее действие, хроническое воздействие паров вызывает острые, а затем хронические риниты, фарингиты, ларингиты, коньюктивиты и бронхиты. |
Свежий воздух; промыть 2% раствором соды нос, глотку, рот, дать теплого молока с содой. При ожоге глаз - длительное промывание водой. |
5 |
III |
|
Уксусный ангидрид |
Сильное раздражающее действие. |
Промыть струей воды. |
20 |
IV |
|
Хлорид -кальция |
При работе вызывает трещины на коже рук. |
Промывание водой кожи рук. |
2,5 |
III |
|
1 |
2 |
3 |
4 |
5 |
|
Хлористый метилен |
Поражает центральную нервную и кроветворную системы, вызывает патологические изменения печени и почек. |
Искуственное дыхание, внутривенно - лобелин, бемегрид; при попадании внутрь - вызвать рвоту, дать слабительное, промывание желудка вазелиновым маслом, инголяция кислородом. |
20 |
IV |
|
Хлористый тионил |
Вызывает раздражение слизистых оболочек дыхательных путей, глаз, легких, кожных покровов. |
Свежий воздух, промывание водой, покой. |
0,5 |
II |
|
Хлороформ |
Наркотический эффект, токсилогическое действие на обмен веществ, внутренние органы, особенно на печень. |
Свежий воздух, искусственное дыхание, покой. |
200 |
IV |
|
Четырех-хлористый углерод |
Наркотик, повреждение печени, почек, легких, потеря сознания, головная боль, головокружение, невриты зрительного нерва. |
Покой, введение глюкозы, исскуственное дыхание, высококалорийная диета |
20 |
II |
|
Этанол |
Действует на центральную нервную и сосудистую системы, печень; наркотик, вызывает тахикардию. |
Промывание желудка, искусственное дыхание |
1000 |
IV |
Таблица 2. Пожароопасные свойства веществ [86,87].
|
Наименование веществ |
Плотность пара по воздуху |
Температура, 0С |
Пределы воспламенения |
|||||
|
Вспышки |
Воспламенения |
Самовоспламенения |
Температурные,0С |
Концентрационные % |
||||
|
нижний |
верхний |
нижний |
верхний |
|||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Ацетон |
2,0 |
-18 |
-5 |
465 |
-20 |
6 |
2,9 |
13 |
|
Ацетонитрил |
1,41 |
6 |
--- |
> 450 |
3 |
--- |
4,1 |
--- |
|
Ацетоуксусный эфир |
--- |
55-72 |
80 |
340 |
40 |
65 |
--- |
--- |
|
Бензол |
2,77 |
-11 |
--- |
634 |
-14 |
13 |
1,4 |
7,1 |
|
Гексан |
3 |
-20 |
-20 |
--- |
-26 |
4 |
1,2 |
7,5 |
|
Гептан |
3,5 |
-4 |
-3 |
202 |
--- |
--- |
1,1 |
6,7 |
|
ДМСО |
1,001 |
87 |
97 |
215 |
--- |
--- |
2,8 |
--- |
|
Диэтиловый. эфир |
2,6 |
-41 |
25 |
164 |
-45 |
13 |
1,7 |
49 |
|
Изопропиловый спирт |
2,1 |
-14 |
--- |
400 |
2 |
12 |
8 |
37 |
|
Метанол |
1.1 |
8 |
13 |
464 |
7 |
39 |
6 |
34,7 |
|
Пиперидин |
2,9 |
16 |
--- |
--- |
--- |
--- |
1,39 |
--- |
|
Толуол |
3,2 |
4 |
--- |
536 |
0 |
30 |
1,3 |
6,7 |
|
Хлористый тионил |
4,1 |
--- |
--- |
555 |
--- |
--- |
--- |
--- |
|
Уксусная кислота |
2,1 |
38 |
--- |
--- |
35 |
76 |
3,3 |
22 |
|
Уксусный ангидрид |
3,5 |
40 |
--- |
360 |
37 |
75 |
1,21 |
9,9 |
|
Хлористый метил |
3 |
14 |
--- |
580 |
--- |
--- |
12 |
22 |
|
Четыреххлористый углерод |
5,3 |
Пары CCl4 оказывают ингибирующее действие на горение многих органических веществ. Минимальная гасительная концентрация-10,5%, т.к. может содержать фосген не применяется в качестве огнегасительного средства. |
||||||
|
Этанол |
1,59 |
16 |
18 |
404 |
11 |
41 |
3,6 |
17,7 |
Таблица 3. Обобщенный анализ потенциальных опасностей.
|
Наименование технологичес-кой операции (ТО) |
Оборудование на котором осуществля-лась ТО |
Реактивы, использовав-шиеся при проведении ТО |
Условия проведения ТО |
Выявление опасности и вредности |
|
Перегонка растворителя |
Электричес-кая плитка, стеклянная посуда, термометр |
Органические растворители |
Зануление электрической плитки |
Поражение электричес-ким током, термический ожог |
|
Проведение реакций |
Электронагреватель, маг-нитная и механическая мешалки, силиконовая баня |
Органические растворители и вещества, кислоты, щелочи |
В вытяж-ном шкафу, ис-пользова-ние термо-реле |
Поражение электричес-ким током, термический или химичес-кий ожог, травмы при работе со стеклом |
|
Фильтрование |
Водоструйный насос, стеклянная посуда |
Органические растворители, вещества |
Вакуум |
Травма стеклом |
|
Хроматография |
Колонка с сорбентом |
Органические растворители, вещества |
--- |
Отравление парами растворителей |
|
Экстракция |
Стеклянная посуда |
Органические растворители, вода, кислоты |
Работа в вытяжном шкафу |
Химические ожоги, травмы стеклом |
Обоснование мер предосторожности при проведении потенциально опасных операций.
1. При взвешивании аналитических эталонов рекомендуется:
обеспечить эффективную вентиляцию;
носить перчатки и лабораторный халат;
принимать меры для предотвращения вдыхания взвешенных частиц и контакта со ртом;
в случае загрязнения следует немедленно промыть кожу, одежду или загрязненную поверхность водой.
2. При работе с ЛВЖ необходимо соблюдать следующие меры предосторожности:
не допускать попадания горючих газов в атмосферу;
при проведении процессов, связанных с нагреванием, пользоваться эффективными водяными холодильниками;
работу проводить в круглодонных колбах из тугоплавкого стекла;
диэтиловый эфир способен при хранении образовывать взрывоопасные перекиси. С целью удаления перекисных соединений проводить очистку растворителя пирогаллолом;
во избежание разлива ЛВЖ и возгорания, большие количества растворителей переливать без плесканий, пользуясь специальной воронкой.
3. При работе с кислотами и щелочами следует пользоваться резиновыми перчатками и защитными очками, так как эти вещества могут вызвать ожоги кожи. При попадании растворов кислот и щелочей на кожу пораженное место нужно промыть струей холодной воды и обработать раствором соды (при попадании кислоты) или борной кислоты (при попадании щелочи).
4. Работа с вакуумом требует соблюдения следующих мер предосторожности:
не использовать в установках плоскодонные колбы и склянки, кроме специально предназначенных для работы при пониженном давлении;
использовать для работы приборы, изготовленные из специального молибденового стекла;
проверять используемую стеклянную посуду на наличие видимых дефектов (трещин, пузырей и др.).
5. В работе использовались различные электроприборы: нагреватели, магнитные мешалки, весы, роторный испаритель.
Основными мерами предотвращения поражений электрическим током в лаборатории являются защита от прикосновения к находящимся под напряжением частям электрооборудования и применение защитного заземления.
Электробезопасность.
Лаборатория кафедры ХТТОС МГАТХТ им. М.В.Ломоносова по классификации помещений по степени опасности поражения электрическим током относится к помещениям без повышенной опасности, так как помещение сухое (с влажностью не более 60%), с нормальной температурой не выше 25о С, с изолированными полами (линолеум) [88,89].
При использовании в лабораторной работе электроустановок запрещалось:
вскрывать электроплитки;
проводить включение электроприборов вблизи ЛВЖ;
применять для подключения электропотребителей проводники с поврежденной изоляцией;
оставлять включенные приборы без присмотра.
При работе в лаборатории использовались следующее электрооборудование: электронагреватели, магнитные и механические мешалки, роторный испаритель, прибор для определения температуры плавления. При их применении контролировались: наличие защитного зануления, наличие заземления, исправность оборудования, соответствие напряжения в сети напряжению для данного прибора.
Оборудование в лаборатории было снабжено защитным занулением, при этом части электроустановок присоединялись к многократно заземленному нулевому проводу, а для снятия статического электричества-заземлением. Была предусмотрена система тройных тумблеров для включения оборудования в электрическую сеть.
Для обеспечения электробезопасности не применялись плитки с открытой спиралью, которые могли бы привести к аварийной ситуации.
Санитарно-гигиенические условия.
Помещение, в котором выполнялась экспериментальная часть магистерской диссертации, характеризовалось малым тепловыделением, работа классифицировалась как легкая [89].
Работа с токсическими и взрывопожароопасными веществами проводилась под тягой, лаборатория была оснащена двумя вытяжными шкафами. Скорость воздуха под тягой в соответствии с паспортными данными составляла не менее 0,7 м/сек., что обеспечивает унос вредных паров с поверхности жидкости. Нормальные санитарно-гигиенические условия в лаборатории обеспечивались нормальной работой приточно-вытяжной вентиляции с кратностью обмена воздуха 2,5.
В лаборатории имелась аптечка медицинская с комплектом средств для оказания первой помощи.
Проводимая в лаборатории работа не требует высокой точности, коэффициент естественной освещенности не менее 1% на самом удаленном от окна рабочем месте. Естественное освещение в лаборатории - боковое. Коэффициент естественной освещенности по СНиП II-4-79 - 1,5. Естественное освещение дополняется искусственным, которое обеспечивается на местах люминесцентными лампами. Освещенность на рабочем месте составляет не менее 100 лк.
Во время выполнения экспериментальной работы образовывались отходы, которые можно разделить на органические и неорганические, жидкие и газообразные. Перед сливом неорганические отходы нейтрализовывали, многократно разбавляли водой и сливали в канализацию. Органические жидкие отходы по возможности регенирировали, перегоняя их, и использовали повторно. Если это было неосуществимо, то отходы собирали в специальные емкости, которые затем уничтожали в общеинститутских масштабах. Условия труда в лаборатории соответствовали санитарным нормам [86,90].
Пожарная опасность в лаборатории и средства пожаротушения.
Необходимые для повседневной работы ЛВЖ и ГЖ (суточная норма не более 1 л) хранились в плотно закрывающихся металлических ящиках, выложенных изнутри асбестом. Концентрированные кислоты и щелочи-в вытяжных шкафах на специальных полках. Реактивы, нестойкие при комнатной температуре, хранились в холодильнике.
Хранение в лаборатории разных групп реактивов требует обязательного соблюдения порядка их совместного хранения.
В соответствии с ОНТП 24-86 лаборатория относится к категории "В", так как работа проводится с большим количеством жидкостей, имеющих температуру вспышки ниже 28 оС, которые не могут, однако, образовывать взрывоопасные концентрации во всем объеме лаборатории [88].
По взрывоопасности в соответствии с ПУЭ лаборатория относится к классу "В-1б", так как имеющиеся в помещении горючие газы и легковоспламеняющиеся жидкости используются в небольших количествах, без применения открытого пламени, и работа с ними проводится в вытяжных шкафах [91].
Группа взрывоопасной смеси паров жидкостей, используемых в работе, по ПИВРЭ соответствует Т1 (температура самовоспламенения горючих веществ более 450 оС.)
Из средств пожаротушения в лаборатории имеются углекислотный огнетушитель, песок, асбестовое одеяло. Из средств индивидуальной защиты в лаборатории есть защитные очки и экраны, резиновые перчатки и фартук, противогаз марки ГП-5, аптечка.
5. Экспериментальная часть.
Для идентификации синтезируемых соединений и контроля протекания реакций использовали ТСХ на пластинах “Silufol UV 254” в различных системах растворителей. Проявляли в парах йода или нагреванием до 120-1400 в течение 2-3 минут или под УФ-лампой. Спектры H1- и C13-ЯМР растворов анализируемых веществ снимали на спектрометрах “Bruker 200SY”(Германия) с рабочей частотой 200Мгц и 50 Мгц соответственно, и “Bruker DPX300”(Германия) с рабочей частотой 300Мгц и 75 Мгц соответственно. ИК-спектры в вазелиновом масле и тонком слое вещества снимали на спектрофотометре “Shimadzu IR 435”(Япония). Масс-спектры снимали на масс-спектрометрах “Finnigan MAT INCOS 50”, метод- электронный удар 70 эВ и “Kratos PC-Kompact MALDI 4”, метод- испарение лазером. Температуру плавления определяли с помощью прибора для определения температуры плавления “Boetius” (Германия). Колоночную хроматографию проводили на силикагеле фирмы Merck Silica gel 60, использовали растворители с характеристиками ХЧ, ОСЧ или Dried.
3–Метил–2,4–пентандион (41).
В трехгорлую колбу на 250 мл, снабженную термометром, обратным холодильником, мешалкой и воронкой для сыпучих веществ, вносят 108 мл абсолютного этанола и добавляют порциями 8,5 г (0,37 моль) Na до растворения. Воронку для сыпучих веществ заменяют на капельную воронку. Образовавшийся этилат натрия нагревают до 450 и добавляют по каплям при той же температуре 38 мл (0,37 моль) 2,4 пентандиона. Полученный раствор охлаждают до 35 400 и прибавляют по каплям 23 мл (0,37 моль) CH3I, таким образом, чтобы температура реакционной массы не превышала 400. Реакционную массу перемешивают в течение 1 часа при 350, затем спирт отгоняют, выпавший осадок NaI растворяют минимальным количеством воды, слои разделяют и водный слой экстрагируют диэтиловым эфиром. Эфирную вытяжку сушат MgSO4. Эфир отгоняют, а остаток перегоняют в вакууме. Получают 20,3 г (48 %) 3 метил 2,4 пентандиона (41).
nD22=1.4440 Ткип=67–680/20–23 мм.
Лит.: nD20=1.4420 Ткип=172–1740 [92].
2–Карбоэтокси–3,4,5–триметилпиррол (43).
1) Изонитрозомалоновый эфир (42).
К раствору 25 мл (0,16 моль) диэтилмалонового эфира в 25 мл уксусной кислоты ксусной кислоты и охлаждении на водяной бане по каплям прибавляют раствор 30 г (0,45 моль) NaNO2 в 40 мл воды при комнатной температуре. Реакционную смесь перемешивают 2 часа при той же температуре. Изонитрозомалоновый эфир (42) отделяют от водного слоя в делительной воронке и без очистки направляют на следующую стадию.
2) 2–карбоэтокси–3,4,5–триметилпиррол (43).
В четырехгорлую колбу на 500 мл, снабженную механической мешалкой, термометром, обратным холодильником и воронкой для сыпучих веществ, помещают раствор 12,8 мл (0,11 моль) 3 метил 2,4 пентандиона (41) в 56 мл уксусной кислоты, одновременно высыпают смесь 24 г (0,36 моль) Zn пыли и 13,5 г (0,17 моль) CH3COONa и нагревают до 900. Воронку для сыпучих веществ заменяют на капельную воронку. В течение 1 часа при постоянном перемешивании и температуре 90–1000 добавляют по каплям раствор 35 мл полученного на предыдущей стадии продукта (42), в 36 мл смеси CH3COOH:H3O (2:1). Реакционную массу выдерживают 1 час при 900 и выливают в 0,5 л ледяной воды. Выпавший осадок отфильтровывают и промывают на фильтре теплой водой. Перекристаллизовывают из метанола. Получают 14,13 г (71%) 2 карбоэтокси 3,4,5 триметилпиррола (43).
Rf =0,7 (Г:ЭА 3:2) ТПЛ = 114–1170 ПМР (CDCl3) (м.д.): 1.32–тр. (3H; CH3–CH3, J=7.2 Гц); 1.89 с. (3H;2–CH3); 2.16–с. (3H; 3–CH3); 2.23–с. (3H; 4–CH3); 4.25–кв. (2H; OCH3, J=7.2 Гц); 8.5 уш.с. (1H,NH).
Лит.: ТПЛ =107–1080 [93]; ТПЛ = 124.5–125.50 [94]; ПМР(CDCl3) (м.д.): 1.35–тр.(3H; CH3 CH3, J=7.1 Гц); 1.89–с.(3H;2–CH3); 2.20–с.(3H; 3–CH3); 2.21–c.(3H; 4–CH3); 4.29–кв.(2H; OCH3, J=7.1 Гц); 9.30–с.(1H; NH). [38]. ИК (neat) (cм 1): 3294, 2992, 2922, 1679, 1441, 1278.[10]
2–Ацетоксиметил–3,4–диметил–5–карбоэтоксипиррол (44).
Тетраацетат свинца.
В трехгорлую колбу на 1 л, снабженную мешалкой и термометром, помещают 408 мл смеси CH3COOH:(CH3CO)2O (5:1) и нагревают до 400, затем прибавляют порциями 137 г (0,2 моль) Pb3O4 так, чтобы температура не превышала 650. Реакционную массу перемешивают при 60–650 до образования прозрачного раствора в течение 2 часов. Затем раствор охлаждают до комнатной температуры, выпавший осадок отфильтровывают. Перекристаллизовывают из смеси CH3COOH:(CH3CO)2O (5:1) и сушат в эксикаторе. Получают 44,5 г (50%) Pb(OAc)4, который направляют на следующую стадию.
2–ацетоксиметил–3,4–диметил–5–карбоэтоксипиррол (44).
В плоскодонной колбе на 100 мл смешивают 30 мл ледяной уксусной кислоты, 1,5 мл уксусного ангидрида и 5 г (0,028 моль) 2 карбоэтокси 3,4,5 триметилпиррола (43), к полученной взвеси добавляют при комнатной температуре 6,1 г (0,014 моль) тетраацетата свинца. Реакционную смесь перемешивают 2 часа до получения прозрачного раствора и выливают в 0,5 л холодной воды. Выпавший осадок отфильтровывают и промывают водой. Перекристаллизовывают из петролейного эфира. Получают 5,38 г (82 %) 2 ацетоксиметил–3,4–диметил–5–карбоэтоксипиррола (44).
Rf = 0,6 (Г:ЭА 3:2) ТПЛ = 98–1000 ПМР (CDCl3) (м.д.): 1.33–тр. (3H; CH3–CH3, J=6.8 Гц); 1.99–с. (3H; CH3CO); 2.05–c. (3H; 3–CH3); 2.23–c. (3H; 4–CH3); 4.28–кв. (2H; OCH3, J=6.8 Гц); 4.99–c. (2H; 2 CH3); 8.94–уш.c. (1H, NH).
Лит.: ТПЛ = 119–1200 [93]; ПМР (CDCl3) (м.д.): 1.12–тр. (3H; CH3–CH3); 1.77–с. (3H; CH3CO); 1.83–с. (3H; 3–CH3); 2.02–с. (3H; 4–CH3); 4.08–кв. (2H; OCH3); 4.80–c. (2H; 2 CH3); 9.12–с. (1H, NH). [93]
1,14-дикарбоэтокси-2,312,13-тетраметилтрипиран (45).
В трехгорлой колбе с обратным холодильником, термометром и прибором для пропускания газов растворяют 7,13 г (0,03 моль) 2-ацетоксиметил-3,4-диметил-5-карбоэтоксипиррола (44) в 175 мл метанола. К полученному раствору прибавляют 1 г (0,015 моль) пиррола и 0,5 г (0,03 моль) толуолсульфокислоты и нагревают раствор до 600 в течение 7 часов, пропуская через него аргон. Реакционную массу охлаждают до комнатной температуры, выпавший осадок отфильтровывают, промывают метанолом и сушат. Получают 3,06 г (51%) 1,14-дикарбоэтокси-2,3,12,13-тетраметил трипирана (45).
Rf = 0,9 (Г:ЭА 1:1) ТПЛ = 146–1500 ПМР (м.д.): 1.29–тр. (6H; CH3–CH3, J=6.8 Гц); 1.96–с. (6H; 2,13-CH3); 2.25–c. (6H; 3,12–CH3); 3.82–c. (4H; –CH3–); 4.26–кв. (4H; OCH3, J=6.8 Гц); 5.81–д. (2H; CH, J=2.56 Гц); 8.65–уш.c. (3H, NH). Масс-спектр m/z(%): 425 (100%).
3,4–Диметил–2–карбоэтоксипиррол.
Натриевая соль 2–метил–3–оксобутилаля.
В колбе, снабженной термометром и воронкой для сыпучих веществ и охлаждаемой смесью льда и соли, смешивают 300 мл сухого диэтилового эфира, 26,9 мл (0,3 моль) метилэтилкетона и 24,2 мл (0,3 моль) этилформиата и охлаждают полученную смесь до –50. Прибавляют порциями 7,6 г (0,33 моль) Na так, чтобы температура не превышала 50. Реакционную массу перемешивают 2 часа на ледяной бане и оставляют на сутки в холодильнике. Выпавший осадок отфильтровывают и промывают холодным диэтиловым эфиром. Получают 28,8 г (78%) натриевой соли 2 метил 3 оксобутилаля, которую без дополнительной очистки направляют на следующую стадию.
3,4–диметил–2–карбоэтоксипиррол.
В четырехгорлую колбу объемом 500 мл, снабженную механической мешалкой, термометром, воронкой для сыпучих веществ и обратным холодильником, помещают 110 мл уксусной кислоты и нагревают до 850 и добавляют 29,4 г ацетата натрия до растворения. Затем последовательно прибавляют 26,9 г (0,22 моль) натриевой соли 2–метил–3–оксобутилаля, 35,5 мл (0,19 моль) изонитрозомалонового эфира (42) и смесь 45 мл CH3COOH и 20 мл H3O и нагревают до 950. К полученному раствору добавляют порциями 41,3 г (0,64 моль) цинковой пыли так, чтобы температура не превышала 1050. Затем реакционную массу перемешивают 0,5 часа при той же температуре и выливают в 0,5 л холодной воды. Экстрагируют хлороформом. Вытяжку сушат MgSO4. Хлороформ отгоняют, а остаток перекристаллизовывают из изопропанола. Получают 2,9 г (10%) 3,4–диметил–2–карбоэтоксипиррола.
Rf = 0,6 (Г:ЭА 3:2); ТПЛ = 90–920; ПМР (м.д.): 1.33–тр. (3H; CH3CH3, J=7.1 Гц); 1.99 с. (3H; CH3); 2.25–c. (3H; CH3); 4.26–кв. (2H; CH3, J=6.8 Гц); 6.63–д. (1H; CH, J=2.98 Гц); 8.75 c. (1H; NH); 13С ЯМР (м.д.): 9.78 (4 CH3); 10.23 (3 CH3); 14.53 (OCH3CH3); 59.77 (О CH3); 119.42 (С-5); 120.41 (С-3, С 4); 126.62 (С-2); 162.02 (С=О). ИК (вазелиновое масло) (cм 1): 3320, 2920, 1656, 1456.
Лит.: ТПЛ = 90–920 [9]; ПМР (CDCl3) (м.д.): 1.33–тр. (3H; CH3CH3, J=7.2 Гц); 1.99–с. (3H; CH3); 2.25–с. (3H; CH3); 4.28–кв. (2H; CH3, J=6.9 Гц); 6.63–д. (1H; CH, J=2.7 Гц); 8.69 с. (1H; NH). [9] 13С ЯМР (CDCl3) (м.д.): 9.84–кв; 10.19–кв; 14.51–кв; 59.73–т; 119.30–c; 120.02–c; 120.51–c; 126.54–д; 161.72–c. [9] ИК (нуйол) (см–1): 3322, 2924, 1731, 1718, 1693, 1671, 1661, 1462. [9]
3,4-диметил-2-карбоксипиррол.
В колбе с обратным холодильником растворяют 2,87 г (0,017 моль) 3,4–диметил–2–карбоэтоксипиррола в 20 мл метанола и прибавляют 10 мл 10% раствора NaOH. Реакционную массу кипятят 3 часа, затем разбавляют водой, охлаждают до комнатной температуры и прикапывают концентрированную HCl до рН=6-7. Выпавший желтый осадок отфильтровывают и сушат на воздухе. Получают 2,3 г (96%) 3,4–диметил–2–карбоксипиррола.
Rf = 0,9 (Г:ЭА 1:1); ТПЛ =2000(разл.); ПМР (CDCl3) (м.д.): 1.98–с. (3H; CH3); 2.24–c. (3H; CH3); 6.63–д. (1H; CH, J=2.6 Гц); 8.63 c. (1H; NH). ИК (вазелиновое масло) (см–1): 3300, 1661.
Лит.: ТПЛ = 2050(разл.) [9]; ПМР (ацетон-d6) (м.д.): 1.93–с. (3H; CH3); 2.20–с. (3H; CH3); 6.71–д. (1H; CH, J=3.0 Гц); 9.30 уш.с. (1H; NH); 10.23-с. (1Н; COOH). [9] 13С ЯМР (ацетон d6) (м.д.): 9.04–кв; 9.43–кв; 118.90–c; 119.44–c; 120.65–c; 125.89–д; 161.85–c. [9] ИК (KBr) (см–1): 3359, 3200-2350, 1648. [9]
1-Бром-2-пропанон (53).
В трехгорлую колбу на 1 литр с обратным холодильником, термометром и капельной воронкой помещают 190 мл воды, 58,6 мл (0,8 моль) ацетона и 44 мл (0,76 моль) уксусной кислоты. При температуре 700 прибавляют по каплям 41 мл (0,8 моль) Br2 до исчезновения окраски, затем раствор перемешивают при той же температуре 0,5 часа и добавляют 100 мл холодной воды. Нейтрализуют твердой содой до pH=6-7 и отделяют органический слой, который сушат над безводным сульфатом магния. Перегоняют в вакууме. Получают 26,3 мл (40%) 1-бром2-пропанона (53).
nD25=1.4670 Ткип=44–490/25–30 мм.
Лит.: nD20=1.4697 Ткип=136,50 [95].
Ацетонилацетоуксусный эфир (54).
1 метод.
В трехгорлой колбе с мешалкой, обратным холодильником и термометром смешивают 200 мл ДМФА, 5,6 г (0,1 моль) мелко измельченного гидроксида калия и 1 каплю катализатора межфазного переноса (аликвотная смесь трикаприлметиламмоний хлорида и триоктилметиламмоний хлорида). При интенсивном перемешивании при комнатной температуре прибавляют по каплям смесь 13 г (0,1 моль) ацетоуксусного эфира и 13,7 г (0,1 моль) 1-бром-2-пропанона (53) с такой скоростью, чтобы температура не превышала 350, после чего смесь перемешивают 1,5 часа при 450. Реакционную массу нейтрализуют 0,5-1,0% раствором HCl до pH=6-7, экстрагируют продукт диэтиловым эфиром. Экстракт сушат MgSO4, эфир отгоняют на роторном испарителе, а остаток перегоняют в вакууме. Получают 3,8 г (20,4%) ацетонилацетоуксусного эфира (54).
nD26,5=1.4370 Ткип=128–1300/20 мм.
Лит.: nD20=1.4375 Ткип=145-1460/ 21 мм. [74], nD20=1.4398 Ткип=87-900/ 0,1 мм. [84].
2 метод.
В трехгорлую колбу на 500 мл с мешалкой, обратным холодильником и термометром помещают 105 мл абсолютного этанола и прибавляют небольшими порциями 6,3 г (0,27 моль) натрия до растворения и образования этилата натрия, затем при комнатной температуре добавляют по каплям 38,4 мл (0,3 моль) ацетоуксусного эфира. Реакционную массу охлаждают до 200 и прикапывают 23 мл (0,27 моль) бромацетона (53), таким образом, чтобы температура не превышала 400, затем температуру повышают до 600 и перемешивают раствор 1 час. Реакционную массу охлаждают до 200, этанол отгоняют на роторном испарителе, в остаток добавляют небольшое количество воды для растворения NaBr. Отделяют нижний слой и экстрагируют из него диэтиловым эфиром. Экстракт сушат с MgSO4. Растворитель отгоняют на роторном испарителе, остаток перегоняют в вакууме. Получают 27 мл (53%) ацетонилацетоуксусного эфира (54).
nD19=1.4382 Ткип=128–1340/20–25 мм.
Лит.: nD20=1.4375 Ткип=145-1460/ 21 мм. [74], nD20=1.4398 Ткип=87-900/ 0,1 мм. [84].
2,5-гександион (55).
Смесь 29,3 г (0,16 моль) ацетонилацетоуксусного эфира (54) и 300 мл 20% раствора K2CO3 в воде кипятят в колбе с обратным холодильником 1 час. Затем раствор охлаждают до комнатной температуры и высаливают K2CO3 до расслаивания. Реакционную массу выливают в делительную воронку и экстрагируют диэтиловым эфиром. Экстракт сушат K2CO3, эфир отгоняют, а остаток перегоняют в вакууме. Получают 11,7 г (65%) 2,5-гександиона (55).
nD22,5=1.4245 Ткип=130–1340/130 мм.
Лит.: nD20=1.4260 Ткип=1910 [96].
2,5-диметилпиррол (56).
В колбе с мешалкой смешивают 6,9 г (0,09 моль)ацетата аммония, 13,4 мл (0,234 моль) уксусной кислоты и 3,4 мл (0,034 моль) уксусного ангидрида при комнатной температуре. К полученному раствору приливают 3 мл (0,026 моль) 2,5-гександиона (55) и перемешивают 30 минут. Реакционную массу нейтрализуют 30% раствором аммиака до рН=7-8 и оставляют на сутки для разложения уксусного ангидрида. Продукт экстрагируют диэтиловым эфиром, экстракт сушат над MgSO4. Эфир отгоняют, а остаток перегоняют в вакууме. Получают 1,4 г (57%) 2,5-диметилпиррола (56).
nD26=1.4780 Ткип=800/20 мм.
Лит.: nD20=1.5066 Ткип=108,30/100 мм.[97].
Нитрование 2,5-диметилпиррола (56).
В трехгорлой колбе с мешалкой, термометром и капельной воронкой охлаждают раствор 1,21 г (0,013 моль) 2,5-диметилпиррола (56) в 7,3 мл уксусного ангидрида до –550. Затем при интенсивном перемешивании прибавляют по каплям смесь 6,1 мл (0,06 моль) уксусного ангидрида и 0,64 мл (0,014 моль) 91,6% HNO3, следя за тем, чтобы температура не превышала –500. Смесь перемешивают 1,5 часа при температуре –550. Затем раствор доводят до 200 и выливают в ледяную воду, нейтрализуют твердой содой. Экстрагируют смесью диэтилового эфира и хлороформа (1:1), экстракт сушат MgSO4. Растворители отгоняют, в остаток добавляют небольшое количество диэтилового эфира, выпавший осадок отфильтровывают. Разделение жидкой фазы проводят с помощью колоночной хроматографии на силикагеле системой элюэнтов с изменяющейся полярностью: этилацетат: петролейный эфир от 1:30 до 1:10. Получают 1,88 г продукта со следующими характеристиками: Rf = 0,2 (Г:ЭА 1:1); ПМР (CDCl3) (м.д.): 2.16–с. (3H); 2.67–c. (2H). 13С ЯМР (CDCl3) (м.д.): 29, 37, 207. ИК (в тонком слое вещества) (см–1): 3475, 3320, 2920, 1714, 1677, 1620.
2,5-диметил-3-карбоэтоксипиррол (63).
В колбе на 250 мл с механической мешалкой смешивают 29 мл (0,17 моль) ацетонилацетоуксусного эфира (54), 38,2 г (0,5 моль) ацетата аммония, 86 мл уксусной кислоты и 22 мл уксусного ангидрида и перемешивают 20 минут. Смесь выливают в 100 мл холодной воды, осадок отфильтровывают. Перекристаллизовывают из изопропанола. Получают 20.3 г (76%) 2,5-диметил-3-карбоэтоксипиррола (63).
Rf = 0,9 (Г:ЭА 1:3); ТПЛ =1130; ПМР (м.д.):1.30-тр. (3H; -CH3CH3; J=7.24 Гц) 2.17–с. (3H; CH3); 2.45–c. (3H; CH3); 4.23–кв. (2H; CH3; J=7.24 ГцРРHHsss); 6.17–д. (1H; CH, J=2.14 Гц); 8.09 уш.c. (1H; NH).
Лит.: ТПЛ = 1130 [79]; ПМР (CDCl3) (м.д.): 1.33–т. (3H; CH3, J=7.2 Гц); 2.19–с. (3H; CH3); 2.47–c. (3H; CH3); 4.25–кв. (2H; CH2, J=7.2 Гц); 6.20–м. (1H; CH); 8.17 уш.с. (1H; NH). [79] ИК (KBr) (см–1): 3293, 1666, 1436, 1224, 1087, 800, 775, 730. [79]
2,5-диметил-3-карбоксипиррол (66).
В колбе с обратным холодильником кипятят смесь 5 г (0,03 моль) 2,5-диметил-3-карбоэтоксипиррола (63), 36 мл метанола и 18 мл 30% раствора NaOH в течение 9 часов. Затем реакционную массу охлаждают и нейтрализуют концентрированной HCl. Выпавший осадок отфильтровывают и сушат на воздухе. Получают 2,7 г (65%) 2,5-диметил-3-карбоксипиррола (66).
Rf = 0,8 (Г:ЭА 1:1); ТПЛ =180-1840 (разл.); ПМР (D2O) (м.д.): 1.92–с. (3H; CH3); 2.21–c. (3H; CH3); 5.81–c. (1H; CH). ИК (вазелиновое масло) (см–1): 3260, 3000-2500, 1639.
2,5-диметил-3,4-дийодпиррол (67).
В трехгорлой колбе с дефлегматором, мешалкой, капельной воронкой и термометром, смешивают 0,5 г (0,0036 моль) 2,5-диметил-3-карбоксипиррола (66) и 20 мл метанола. При перемешивании в полученную суспензию прикапывают раствор 1,24 г (0,009 моль) K2CO3 в 20 мл воды и нагревают до 650, при этой температуре прибавляют раствор 0,76 г (0,003 моль) I2 и 1,15 г (0,007 моль) KI в 10 мл воды. Реакционную массу перемешивают 20 минут и охлаждают. Осадок отфильтровывают, промывают водой и перекристаллизовывают из метанола. Получают 0,56 г (45%) 2,5-диметил-3,4-дийодпиррола (67).
Rf = 0,8 (Г:ЭА 1:3); ТПЛ =116-1200 (разл.); ПМР (м.д.): 2.28–с. (6H; CH3); 8.01 уш.c. (1H; NH). Масс спектр m/z (%): 347 (100%).
2,5-диметил-3-иод-4-карбоэтоксипиррол (64).
В трехгорлой колбе с дефлегматором, мешалкой, капельной воронкой и термометром, растворяют 0,5 г (0,003 моль) 2,5-диметил-3-карбоэтоксипиррола (63) в 20 мл метанола. При перемешивании в полученный раствор прикапывают раствор 1,24 г (0,009 моль) K2CO3 в 20 мл воды и нагревают до 650, при этой температуре прибавляют раствор 0,76 г (0,003 моль) I2 и 1,15 г (0,007 моль) KI в 10 мл воды. Реакционную массу перемешивают 20 минут и охлаждают. Осадок отфильтровывают и перекристаллизовывают из метанола. Получают 0,69 г (80%) 2,5-диметил-3-иод-4-карбоэтоксипиррола (64).
Rf = 0,7 (Г:ЭА 1:1); ТПЛ =116-1200;
ПМР (CDCl3) (м.д.):1.34-тр. (3H; -CH3CH3; J=7.24 Гц) 2.22–с. (3H; CH3); 2.47–c. (3H; CH3); 4.26–кв. (2H; CH3; J=7.24 ГцРРHHsss); 8.18 уш.c. (1H; NH). 13С ЯМР (CDCl3) (м.д.): 13.89 (2-CH3); 14.19 (3-CH3); 14.30 (OCH2CH3); 50.46 (C-4); 59.58 (C-I); 63.03 (-CH2-); 129.48 (C-5); 135.58 (C-2); 164.62 (C=O). ИК(вазелиновое масло) (см 1): 3256, 1675, 1217, 1099, 1029, 773. Масс спектр m/z (%): 293 (40%), 279 (20%), 264 (42%), 248 (28%), 219 (8%), 127 (12%),122 (25%), 93 (30%), 67 (35%), 51 (55%), 42 (100%).
2,5-диметил-3-карбоэтокси-4-нитропиррол (65).
1метод.
В колбе на 25мл с мешалкой растворяют 0.5г (0.003 моль) 2,5-диметил-3-карбоэтоксипиррола (63) в 10мл ацетонитрила. При перемешивании в полученный раствор при комнатной температуре приливают раствор 0.6г (0.004 моль) AgNO2 в 2мл ацетонитрила, затем при этой же температуре прибавляют раствор 0.5г(0.002 моль) I2 в 8мл ацетонитрила. Реакцию проводят в течение 48ч при комнатной температуре в инертной атмосфере в темноте. Выпавший осадок отфильтровывают, разделение жидкой фазы проводят при помощи колоночной хроматографиии на силикагеле в системе элюэнтов: ЭА:ПЭ (1:3). Получают 0.11г (18%) 2,5-диметил-3-карбоэтокси-4-нитропиррола (65).
Rf =0.6 (ПЭ:ЭА 1:3); Тпл= 110-1130 ; ПМР(CDCl3) (м.д.): 1.32 м.д.-тр.(3Н, СН2 СН3, J=7.2 Гц); 2.35 м.д.-с.(3Н, СН3 ); 2.48 м.д.-с.(3Н, СН3); 4.30 м.д.-кв.(2Н, СН2-СН3, J=7.2 Гц); 8.91 м.д.-уш.с.(1Н, NH). ИК(вазелиновое масло) (см–1) : 3317, 1720, 1680, 1600, 1110, 1020. Масс-спектр m/z(%): 212 (19%), 166 (40%), 122 (53%), 92 (45%), 65 (22%), 51 (16%), 42 (100%).
2 метод.
В колбу на 15мл с обратным холодильником вносят раствор 0.4г(0.001 моль) 2,5-диметил-3-иод-4-карбоэтоксипиррола (64) в 5мл ацетонитрила и добавляют к нему раствор 0.42г (0.003 моль) AgNO2 в 3мл ацетонитрила. Реакционную массу кипятят 2ч при t бани =110 и оставляют на 2 суток стоять при комнатной температуре в темноте. Выпавший осадок отфильтровывают, продукт очищают с помощью колоночной хроматографии насиликагеле(l=17см,d=20см) в системе растворителей диэтиловый эфир: петролейный эфир (1:2). Получают 0.1г (47%) 2,5-диметил-3-карбоэтокси-4-нитропиррола (65).
Rf =0.5(ПЭ:ЭА 1:1); Тпл= 110-1120 ; ПМР(CDCl3) (м.д.): 1.35 м.д.-тр.(3Н, СН2 СН3, J=7.1 Гц); 2.35 м.д.-с.(3Н, СН3 ); 2.48 м.д.-с.(3Н, СН3); 4.32 м.д.-кв.(2Н, СН2-СН3, J=7.1 Гц); 8.91 м.д.-уш.с.(1Н, NH). ИК(вазелиновое масло) (см–1) : 3300, 1720, 1680, 1600. Масс-спектр m/z(%): 212 (19%), 166 (40%), 122 (43%), 92 (45%), 66 (28%), 54 (19%), 42 (100%).
2-формилпиррол (58).
В колбе, снабженной термометром, мешалкой и капельной воронкой и охлаждаемой смесью льда и соли, при температуре 0-50 к 45 мл ДМФА прикапывают 15,4 мл POCl3. К полученному формилирующему комплексу при температуре не выше 150 прибавляют 10,36 мл (0,15 моль) пиррола (57), после чего повышают температуру до 350 и перемешивают 0,5 часа. Реакционную массу выливают в 150 мл холодной воды, из полученного раствора экстрагируют диэтиловым эфиром следы не прореагировавшего пиррола (57). Водный слой обрабатывают 150 мл насыщенного раствора ацетата натрия и кипятят 0,5 часа с обратным холодильником. Раствор охлаждают и экстрагируют дихлорметаном, экстракт промывают несколько раз водой, удаляя ДМФА. Экстракт сушат сульфатом натрия, растворитель отгоняют, а остаток перегоняют в вакууме. Получают 14,25 г (64%) 2-формилпиррола (58).
Rf = 0,6 (Г:ЭА 3:2); ТПЛ = 400; ИК (в тонком слое ): 3260; 1650.
Лит.: ТПЛ =41-440 [83]; ПМР (ацетон-d6) (м.д.): 6.3–с. (1H; 4-CH); 7.0–д. (1H; 3-CH); 7.3 с. (1H; 5-CH) [83]; ИК(CCl4) (см–1): 3450(NH); 1665(C=O); 1655. [83]
2,5-диформилпиррол (61).
Диэтил 2-(пиррол-2-илметилен)малоноат (59).
В круглодонной колбе на 25 мл с насадкой Дина-Старка кипятят смесь 0,3 г (0,0032 моль) 2 формилпиррола (58), 0,5 г (0,0032 моль) диэтилмалонового эфира, 0,04 г (0,00063 моль) уксусной кислоты и 0,01 г (0,00013 моль) пиперидина в 9 мл бензола в течение 1 часа. Реакционную массу охлаждают до комнатной температуры и промывают полунасыщенным раствором хлорида натрия. Бензольный слой сушат с безводным сульфатом натрия, растворитель отгоняют на роторном испарителе. Получают 0,73 г (97%) диэтил 2-(пиррол-2-илметилен) малоноата (59), который без дополнительной очистки направляют на следующую стадию.
Диэтил 2-[(5-формилпиррол-2-ил)метилен]малоноат (60).
В колбе, снабженной термометром и капельной воронкой и охлаждаемой смесью льда и соли, при температуре 0-50 в атмосфере аргона к 0,3 мл ДМФА прикапывают 0,27 мл POCl3. К полученному формилирующему комплексу при температуре не выше 150 прибавляют 0,63 г (0,0026 моль) диэтил 2-(пиррол-2-илметилен)малоноата (59), после чего повышают температуру до 400 и перемешивают 0,5 часа. Реакционную массу охлаждают, разбавляют хлороформом и промывают водой. Органический слой сушат с безводным сульфатом натрия, растворитель отгоняют на роторном испарителе. Получают 0,38 г (55%) диэтил 2 [(5 формилпиррол-2-ил)метилен]малоноата (60), который без дополнительной очистки направляют на следующую стадию.
Характеристики очищенного с помощью колоночной хроматографии (d =1.5 см; l = 20 см) на силикагеле (элюэнт хлороформ: метанол 9:1) продукта: Rf = 0,7 (Хл:МеОН 9:1); ТПЛ = 98–1000; ПМР (CDCl3) (м.д.): 1.31–тр. (3H; CH3CH3, J=7.26 Гц); 1.37–тр. (3H; CH3CH3, J=7.26 Гц); 4.30–кв. (2H; CH3, J=7.26 Гц); 4.38–кв. (2H; CH3, J=7.26 Гц); 6.68–д.д. (1H; CH, J=2.14 Гц); 6.94–д.д. (1H; CH, J=2.14 Гц); 7.57–c. (-CH=C); 9.65 с. (CHO); 11.53 уш.c. (1H; NH); 13С ЯМР (м.д.): 13.88 (OCH3CH3); 13.99 (OCH3CH3’); 61.60 (О CH3); 62.11 (О CH3’); 119.53 (=С-); 120.49 (С 4); 121.92 (С-3); 131.85 (C-5); 134.09 (-C=); 135.63 (C-2); 163.30 (С=О); 166.66 (С=О’); 179.78 (CHO). ИК (вазелиновое масло) (cм 1): 3300, 1730, 1700, 1670, 1620, 1550. Масс-спектр m/z (%): 265 (71%), 173 (100%), 145 (46%), 119 (30%), 91 (28%), 65 (30%), 39(15%).
2,5-диформилпиррол (61).
В колбе с дефлегматором кипятят 0,56 г (0,0021 моль) диэтил 2 [(5 формилпиррол-2-ил)метилен]малоноата (60) в 30 мл 3М раствора NaOH в течение 1 часа. Потемневший раствор охлаждают и нейтрализуют разбавленной серной кислотой до pH=6-7, выпавший коричневый осадок отфильтровывают, промывают водой и сушат в вакууме. Получают 0,25 г (96%) 2,5-диформилпиррола.
Rf = 0,3 (Хл:МеОН 9:1); ТПЛ = 112–1140; ПМР (CDCl3) (м.д.): 7.02–д. (2H; CH, J=2 Гц); 9.77 с. (2H; CHO); 10.38 уш.c. (1H; NH); ИК (вазелиновое масло) (cм 1): 3140, 1720, 1700. Масс-спектр m/z (%): 123 (65%), 94 (18%), 66 (60%), 39(100%).
Нитрование 2,5-диформилпиррола (61).
В колбе на 25мл с мешалкой растворяют 0.05г (0.0004моль) 2,5-диформилпиррола (61) в растворе 4мл (0.07моль) уксусной кислоты и 2мл (0.02моль) уксусного ангидрида. При перемешивании в полученный раствор прибавляют смесь 0.022мл (0.0005моль) HNO3(98%) и 0.136мл (0.0014моль) уксусного ангидрида. Реакционную смесь перемешивают 2ч при комнатной температуре, а затем прибавляют еще0.022мл (0.0005моль) HNO3(98%) и перемешивают 0.5ч. Далее реакционную массу выливают на небольшое количество льда, нейтрализуют твердой содой до рН= 6-7, приливают 20мл толуола и упаривают азеотропную смесь на роторном испарителе. Затем выпавший осадок промывают ацетоном и упаривают растворитель на роторном испарителе. Получают 0.007г (10%) 2-карбокси-5-формилпиррола (62).
Rf = 0,4(Ме); ТПЛ = 112–1140, ПМР (CDCl3-CD3OD) (м.д.): 6.15–д. (1H; CH, J=4 Гц); 6.68–д. (1H; CH, J=4 Гц); 7.94 с. (1H; CHO); ИК (вазелиновое масло) (cм 1): 3450, 1720, 1700,1620,1600,1530,1380,1300.
Глицинэтилового эфира хлоргидрат (49).
К суспензии 10,2 г (0,135 моль) глицина (48) в 100 мл 96% этанола прибавляют по каплям 14,6 мл (0,2 моль) тионилхлорида с такой скоростью, чтобы реакционная смесь слабо кипела. Смесь кипятят с обратным холодильником 2 часа. Затем растворитель отгоняют на роторном испарителе, а остаток растворяют при нагревании в 60 мл 96% этанола и охлаждают, выпавшие белые хлопья отфильтровывают и промывают диэтиловым эфиром. Получают 17,8 г (94%) хлоргидрата глицинэтилового эфира (49).
ТПЛ = 138-1400.
Лит.: ТПЛ =145-1460 [16]; ПМР (DMSO-D6/ CDCl3) (м.д.): 1.27–т. (3Н; CH3, J=7 Гц); 3.73–с. (2Н; СH2-N); 4.23–кв. (2H; CH2, J=7Гц); 8.55-уш.с. (3H; NH) [16]; ИК (KBr) (см–1): 3330-2300 ( +NH); 1745(C=O), 1250, 1050, 990. [16]
N-формилглицина этиловый эфир (50).
В колбе с обратным холодильником и капельной воронкой смешивают 25 мл (0,37 моль) этилортоформиата, 13 мг п-толуолсульфокислоты и 17,8 г (0,13 моль) хлоргидрата глицинэтилового эфира (49) и нагревают до кипения. В полученную смесь прикапывают 19,5 мл (0,14 моль) триэтиламина и кипятят 20 часов. Затем реакционную массу охлаждают до комнатной температуры, выпавший осадок гидрохлорида триэтиламина отфильтровывают. Фильтрат упаривают на ¾ и охлаждают до –50, выпавший гидрохлорид триэтиламина снова отфильтровывают, а фильтрат перегоняют в вакууме. Получают 9,5 мл (66%) этилового эфира N формилглицина (50).
nD19,5=1.4510 Ткип=81-900/0,05 мм.
Лит.: nD20=1.4530 Ткип=1100/0,1 мм.[16].
Этиловый эфир изоциануксусной кислоты (51).
В колбе, снабженной термометром, мешалкой и капельной воронкой и охлаждаемой смесью льда и соли, при температуре 00 к раствору 10,93 г (0,083 моль) этилового эфира N формилглицина (50) и 29 мл (0,21 моль) триэтиламина в 85 мл дихлорметана прикапывают 12,8 г (0,083 моль) POCl3 и смесь перемешивают 1 час при этой температуре. Затем медленно прибавляют при 20-250 раствор 16,7 г Na2CO3 в 80 мл воды, соблюдая указанный интервал температур и полученную смесь перемешивают 30 минут при этой температуре. Органическую фазу отделяют, а из водного слоя экстрагируют дихлорметаном. Объединенные органические фазы промывают насыщенным раствором NaCl и сушат над K2CO3. После отгонки растворителя остаток перегоняют в вакууме. Получают 6,21 г (66%) этилового эфира изоциануксусной кислоты (51).
nD20=1.4175 Ткип=85-860/15 мм. ПМР (CDCl3) (м.д.): 1.25–т. (3Н; CH3, J=7.16 Гц); 4.20–с. (2Н; СH2-N); 4.24–кв. (2H; CH2, J=7.16 Гц).
Лит.: Ткип=80-820/12 мм.[16]. ПМР (CCl4) (м.д.): 1.33–т. (3Н; CH3, J=7 Гц); 4.25–с. (2Н; СH2-N); 4.28–кв. (2H; CH2, J=7 Гц) [16]; ИК (пленка) (см–1): 2150 (NС); 1750(C=O) [16].
2-метил-6-нитробензотиазол (47).
В колбе с дефлегматором готовят нитрующую смесь из 0,7 мл 73% HNO3 и 0,83 мл концентрированной H2SO4 к которой прикапывают 1 мл (0,008 моль) 2-метилбензотиазола (46). Реакционную массу греют при 900 в течение 5 часов и выливают на лед. Выпавший осадок отфильтровывают и промывают большим количеством воды. Перекристаллизовывают из метанола. Получают 0,22 г (20%) 2-метил-6-нитробензотиазола (47).
Rf = 0,9 (Хл:МеОН 9:1); ТПЛ = 106–1100; ПМР (м.д.): 2.9–с. (3H; CH3); 8.03–.д. (1H; CH, J=8.96 Гц); 8.35–д.д. (1H; CH, J=2.29 Гц); 8.78–д. (1H; CH, J=2.29Гц). Масс-спектр m/z (%): 194 (94%), 164 (48%), 148 (50%), 136 (30%), 107 (35%), 69 (30%), 63(100%).
6. Выводы.
Проведен анализ литературы и подобраны методы получения высоко сопряженных порфиринов.
Предложен новый метод синтеза 2,5-диформилпиррола.
Разработаны методы синтеза 5 новых соединений: диэтил 2-(пиррол-2-илметилен)малоноат (59), диэтил 2 [(5 формилпиррол-2-ил)метилен]малоноат (60), 2,5-диметил-3-йод-4-карбоэтоксипиррол (64), 2,5-диметил-3-карбоэтокси-4-нитропиррол (65), 2,5-диметил-3,4-дийодпиррол (67).
7. Литература.
Lash T.D. and Novak B.H. New highly conjugated porphyrin chromophores: synthesis of mono- and diphenanthroporphyrins. // Tetrahedron Letters 1995 v.36 №25 p.4381-4384.
Alonso C.M.A., Neves M., et al. Reaction of -amino-meso-tetraphenylporphyrin with ,-unsaturated carbonyl compounds: an approach to fused pyridinoporphyrins. // Tetrahedron Letters 1997 v.38 №15 p.2757-2758.
Vicente M., Jaquinod L., Khoury R., Mandrona A., Smith K.M. Synthesis and chemistry of new benzoporphyrins. // Tetrahedron Letters 1999 v.40 p.8763-8766.
Silva A., Faustino M., Silva T., et al. A new approach to the synthesis of mono- and bis-pyrroloporphyrins. // Abstracts of ICPP-1, Dijon, France, 2000, post 402.
Lin Y. and Lash T.D. Porphyrin synthesis by the “3+1” methodology: a superior approach for the preparation of porphyrins with fused 9.10-phenanthroline sub>units. // Tetrahedron Letters 1995 v.36 p.9441-9444.
Novak B.H. and Lash T.D. Porphyrins with exocyclic rings. Part 11. Synthesis and characterization of phenanthroporphyrins, a new class of modified porphyrin chromophores. // J.Org.Chem. 1998 v.63 p.3998-4010.
Lash T.D., Wijesinghe C., Osuma A.T., Patel J.R. Synthesis of novel porphyrin chromophores from nitroarenes: further applications of the Barton-Zard pyrrole condensation. // Tetrahedron Letters 1997 v.38 №12 p.2031-2034.
Lash T.D. Porphyrins with exocyclic rings. Part 10. Synthesis of meso,-propanoporphyrins from 4,5,6,7-tetrahydro-1H-indoles. // Tetrahedron 1998 v.54 p.359-374.
Byun Y.-S. And Lightner D.A. Synthesis and properties of a bilirubin analog with propionic acid groups replaced by carboxyl. //J.Heterocycl. Chem. 1991 v.28 № 7 p.1683-1692.
Cho D.H., Lee J.H., Kim B.H. An improved synthesis of 1,4-bis(3,4-dimethyl-5-formyl-2-pyrryl)butadiyne and 1,2-bis(3,4-dimethyl-5-formyl-2-pyrryl)ethyne. // J.Org.Chem. 1999 v.64 p.8048-8050.
Piloty O., Hirsch P. Pyrrolsynthesen aus Aminoketonen mit ketonen und ketonsaureestern. //J.Liebigs Ann.Chem. 1913 B.395 s.63-74.
Treibs A., Zinsmeister R., Schmidt R. Uber die Knorrschepyrrolsynthese. //Chem.Ber. 1957 B.90 s.79-84.
Johnson A.W., Price R. 2,3,4,5-Tetramethylpyrrole. //Org. Synthesis 1962 v.42 p.90 92.
Treibs A., Schmidt R. Syntheische Arbeiten auf dem chlorophyllgebiet synthese des 2 Desathylphylloporphyrins. //J.Liebigs Ann.Chem. 1952 B.577 s.105-115.
All G.H., Knowles W.S. The mechanism of the N,N-dichloroalkylamine rearrangement. //J.Org.Chem. 1960 v.25 p.2047-2048.
Титце Л., Айхер Т. //Препаративная органическая химия. Пер. с нем. под ред. Алексеева Ю.Е. М.: Мир, 1999. 704 с.
Миронов А.Ф., Апаркон Х.Х., Евстигнеева Р.П. О лабильности диэтиламиноэтильной группы в условиях образования пиррольного цикла по Кнорру. //ХГС 1973 №12 стр.1643-1645.
Nagafuji P. and Cushman M. A general synthesis of pyrroles and fused pyrrole systems from ketones and amino acids. //J.Org.Chem. 1996 v.61 №15 p.4999-5003.
Barret A.G.M., Graboski G.G. Conjugated nitroalkenes: versatile intermediates in organic synthesis. //Chem.Rev. 1986 v.86 №5 p.751-762.
Chandrasekar P. and Lash T.D. Versatile “3+1” syntheses of acenaphthoporphyrins, a new family of highly conjugated tetrapyrroles. // Tetrahedron Letters 1996 v.37 №28 p.4873 4876.
Murashima T., Tamai R., Fujita K., Uno H. and Ono N. Ambident reactivity of nitro heteroaromatic anions. // Tetrahedron Letters 1996 v.37 №46 p.8391-8394.
Fumoto Y., Uno H., Ono N., et al. Preparation of 5-unsub>stituted 4-formylpyrrole-2-carboxylates and conversion to cycloalkano-oligopyrroles. // J.Chem.Soc.PerkinTrans.1. 2000 p.2977-2981.
Gilchrist T.L. Synthesis of aromatic heterocycles. //J.Chem.Soc.PerkinTrans.1. 1998 №3 p.615-628.
Chiu P. -K., Lui K. -H., Maini P.N. Sammes M.P. Novel synthesis of 3H-pyrroles, and novel intermediates in the Paal-Knorr 1H-pyrrole synthesis: 2 hydroxy 3,4 dihydro 2H pyrroles from 1,4-diketones and liquid ammonia. //J.Chem.Soc.,Chem.Commun. 1987 p.109-110.
Niziurski-Mann R.E. and Cava M.P. Synthesis of mixed thiophene-pyrrole heterocycles. //J.Heterocycles 1992 v.34 №10 p.2000-2021.
Benary E. Synthese von pyrrol- und furan-derivaten aus dichlor-ather, acetessigester und ammoniak. //Chem.Ber. 1911 B.44 s. 493-496.
Khotinsky E. Darstellung des pyrrols. //Chem.Ber. 1909 B.9 s.2506-2507.
Chiu P. -K. and Sammes M.P. The synthesis and chemistry of azolenines. Part 18. Preparation of 3-etoxycarbonyl 3H pyrroles via the Paal Knorr reaction, and sigmatropic rearrangements involving competitive ester migrations to C 2, C 4 and N. //Tetrahedron 1990 v.46 №10 p.3439-3456.
Rigo B., Valligny D., Taisne S., Couturier D. Disilylated compounds as precursors of heterocycles. //J.Synth.Commun. 1988 v.18 p.170-171.
Hendrickson J.B., Ress R.W., Templeton J.F. General heterocycle synthesis. Use of acetyl enedicarboxylic esters. //J.Am.Chem.Soc. 1964 v.86 p.107-111.
Grob C.A., Schacl H.P. Eine nene pyrroling- synthese II. Teil untersuchungen inder pyrrolreihe. //Helv.Chim.Acta 1955 v.38 p.1121-1127.
Spence J.D. and Lash T.D. Porphyrins with exocyclic rings. Part 14. Synthesis of tetraacenaphthoporphyrins, a new family of highly conjugated porphyrins with record breaking long wavelengh electronic absorptions. //J.Org.Chem. 2000 v.65 p.1530-1539.
Bastian J.A. and Lash T.D. Porphyrins with exocyclic rings. Part 12. Synthesis of meso, butano- and meso, pentanoporphyrins from cycloalka[b]pyrrole. //Tetrahedron 1998 v.54 p.6299-6310.
Gotthardt H., Huisgen R. And Bayer H.O. 1.3-Dipolar cycloaddition reactions. L III. The question of the 1.3-dipolar nature of 2-oxazolin-5-ones. //J.Am.Chem.Soc. 1970 v.92 p.4340 4343.
Arcadi A. and Rossi E. Synthesis of functionalised furans and pyrroles through annulation reactions of 4-pentynones. //Tetrahedron 1998 v.54 p.15253-15272.
Sessler J.L., Davis J.M., Lynch V. Synthesis and characterization of a stable smaragdyrin isomer. //J.Org.Chem. 1998 v.63 p.7062-7065.
Alberola A., Ortega A.G., et.al. Versatility of Weinreb amides in the Knorr pyrrole synthesis. //Tetrahedron 1999 v.55 p.6555-6566.
Hombrecher H.K., Horter G. Synthesis of pyrroles via ethyl N- (3 oxo 1 alkenyl) glycinates. //Synthesis 1990 p.389-391.
Ferraz H.M.C., Oliveira E.O., et.al. A new and efficient approach to cyclic enamino esters and enamino ketones by iodine- promoted cyclization. //J.Org.Chem. 1995 v.60 p.7357-7359.
Ferraz H.M.C., Pereira F.L.C., et.al. Synthesis of N sub>stituted pyrrole and tetrahydroindole derivatives from alkenyl dicarbonyl compounds. //Tetrahedron 1999 v.55 p.10915 10924.
Рындина С.А., Кадушкин А.В., Соловьева Н.П., Граник В.Г. Циклизация Торпа Циглера в синтезе 3 амино 4 цианопиррола. //ХГС 2000 т.26 с.1643-1655.
Chen N., Lu Y., Gadamasetti K., et.al. A short, facile synthesis of 5-sub>stituted 3 amino 1H pyrrole 2 carboxylates. //J.Org.Chem. 2000 v.65 p.2603-2605.
Порфирины: структура, свойства, синтез. // Под ред. Ениколопяна Н.С. М.: Наука, 1985. 333с.
Rose E., Soleihavoup M., et al. Bis-faced aminoporphyrin templates for the synthesis of chiral catalysts and hemeprotein analogues. // J.Org.Chem. 1998 v.63 №6 p.2042-2044.
Ono N., Muratani E., et al. Synthesis of 2,7,12,17 tetraaryl-3,8,13,18-tetranitroporphyrins; electronic effects on aggregations of porphyrins. // J.Chem.Soc., Perkin Trans.1 1998 №22 p.3819-3824.
Tse M.K., Zhou Z., et al. Regioselective bromination and sub>sequent suzuki cross-coupling of highly electron deficient 5,10,15,20-tetrakis(trifluoromethyl)porphyrin. // Tetrahedron 2000 v.56 p.7779-7783.
Czuchajonski L., Habdas J., et al. Porphyrinyl-uridines as the first water soluble porphyrinyl-nucleosides. // Tetrahedron Letters 1991 v.32 p.7511-7514.
Shin J.-Y., Minezawa N., et al. Study for expanded porphyrins producted during the condensation reaction of pentafluorobenzaldehyde and pyrrole. // Abstracts of ICPP-1, Dijon, France, 2000, post 562.
Ono N. A new synthesis of highly conjugated porphyrins. // Abstracts of ICPP-1, Dijon, France, 2000, sym 147.
Ono N., Hironaga H., et al. A new synthesis of pyrroles and porphyrins fused with aromatic rings. // J.Chem.Soc., Perkin Trans.1 1996 p.417-423.
Nguyen L.T., Senge M.O., Smith K.M. One-pot synthesis of regiochemically pure porphyrins from two different pyrroles. // Tetrahedron Letters 1994 v.35 p.7581-7584.
Nguyen L.T., Senge M.O., Smith K.M. Simple methology for syntheses of porphyrins possessing multiple peripheral sub>stituents with an element of symmetry. // J.Org.Chem. 1996 v.61 p.998-1003.
Березин Б.Д. // Координационные соединения порфиринов и фталоцианина / М.: Наука 1978 150с.
Arsenault G.P., Bullock E., MacDonald S.F. Pyrromethanes and porphyrins there from. // J.Am.Chem.Soc. 1960 v.82 p.4384-4387.
Clarke O.J., Boyle R.W. Selective synthesis of asymmetrically sub>stituted 5,15 diphenylporphyrins. // Tetrahedron Letters 1998 v.39 p.7167-7168.
Lee C.-H., Li F., Iwamoto K., Lindsey J.S. Synthetic approaches to regioisomerically pure porphyrins bearing four different meso-sub>stituents. // Tetrahedron 1995 v.51 p.11645-11654.
Balasub>ramanian T., Lindsey J.S. synthesis of -sub>stituted porphyrin building blocks and conversion to diphenylethyne-linked porphyrin dimers. // Tetrahedron 1999 v.55 p.6771 6784.
Maruyama K., Nagata T., Ono N., Osuka A. A synthesis of unsymmetric porphyrin dimers // Bull.Chem. Soc.Jpn. 1989 v.62 p.3167-3170.
Ema T., Kuroda Y., Ogoshi H. Selective syntheses of unsymmetrical meso-arylporphyrins. // Tetrahedron Letters 1991 v.32 p.4529-4532.
Wallaca D.M., Leung S.H., Senge M.O., Smith K.M. Rational tetraarylporphyrin syntheses: tetararylporphyrins from the MacDonald route. // J.Org.Chem. 1993 v.58 p.7245-7257.
Мамардашвили Н.Ж., Голубчиков О.А. Синтез порфиринов из дипирролилметанов. // Успехи химии 2000 т.69 с.342-354.
Scog W.R., Yong H.N., Youngkyu D. Synthesis, structures and electrchemical characterization of ferrocene-sub>stituted porphyrin and porphodimethene. // Inorg.Chim.Acta 2000 v.309 p.49-56.
Tjahjono D.H., Akutsu T., et al. Cationic porphyrins bearing diazolium rings: synthesis and their interaction with calf thymus DNA. // Biochimica et Biophysica Acta/General sub>jects 1999 v..1472 p.333-343.
Smith K.M., Craig J., Medforth D.T.L. Syntheses, stability and tumorcidal activity of porphyrin dimers and trimers with ether linkages. // Tetrahedron Letters 1990 v.31 p.7265-7270.
Khoury R.G., jaquinod L., Smith K.M. Metal ion-induced self assembly of open-chain tetrapyrrole derivatives: double stranded dinuclear complexes from 10-oxo-5,15-biladienes. // Tetrahedron 1998 v.54 p.2339-2346.
Dolphin D.M., Johnson A.W., Long J. Porphyrinogens and porphodimethenes, intermediates in the synthesis of meso-tetraphenylporphyrins from pyrroles and benzaldehyde. // J.Heterocycl.Chem. 1970 v.7 p.275-283.
Smith K.M., Minnetian O.M. Anomalous cyclization of 1,19-dimethyl-a,c-dimethyl-a,c-biladiens: direct synthesis of meso-aminoporphyrin derivatives. // Synth.Commun. 1985 v.15 p.75-80.
Smith K.M., Minnetian O.M. Cyclization of 1’,8’-dimethyl-a,c-biladiene salts to give porphyrins: a study with various oxidizing agents. // J.Chem.Soc.,Perkin Trans.1 1986 p.277 280.
Boudif A., Gimenez S., Loock B., Momenteau M. vic-Diacrylic ester porphyrins as starting materials for monobenzoporphyrins and opp-dibenzoporphyrins syntheses. // Can.J.Chem. 1998 v.76 p.1215-1219.
Boudif A., Momenteau M. A new convergent method for porphyrin synthesis based on a “3+1” condensation. // J.Chem.Soc.,Perkin Trans.1 1996 p.1235-1241.
Kai S., Suzuki M., Masaki Y. The first synthesis of mononazaporphyrins bearing a nitrogen atom at the peripheral position. // Tetrahedron Letters 1998 v.39 p.4063-4066.
Lash T.D., Chandrasekar P., et al. Porphyrins with exocyclic rings. Part 13. Synthesis and spectroscopic characterization of highly modified porphyrin chromophores with fused acenaphthylene and benzothiadiazole. // J.Org.Chem. 1998 v.63 p.8455-8469.
Lash T.D., Thompson M.L., et al. Recent studies on the synthesis of porphyrins with fused aromatic rings. // Abstracts of ICPP-1, Dijon, France, 2000, post 469.
Сизов А.Ю., Яновская Л.А., Домбровский В.А. Синтез эфиров 2-замещенных 4-кетопентановых кислот алкилированием СН-кислот хлорацетоном в условиях межфазного катализа. // Известия РАН 1990 №2 с.473-474.
Cresp T.M., Sargent M.V. Synthesis and paratropicity of heteroatom-bridged annulenones.//J.Chem.Soc., Perkin Trans.1.1973.N.23.P.2961-2971.
Muchowski J.M. and Hess P. Lithiation of the 6-dimethylamino-1-azafulvene dimer. A versatile synthesis of 5-sub>stituted pyrrole-2-carboxaldehydes. //J.Tetrahedron Lett.1988.V.29.N26.P.777-780.
Muchowski J.M. and Hess P. Lithiation of the dimer of 3-bromo-6-dimethylamino-1-azafulvene. Efficacious synthesis of 4-mono- and 4,5-disub>stituted pyrrole-2-carboxaldehydes.//J.Tetrahedron Lett.1988.V.29.P. 3215-3219.
Bergman J., Renstroem L., Sjoerberg B. The synthesis of pyrrole-2.5-dicarbaldehydes.// J.Tetrahedron. 1980. V.36. P.2505-2509.
Muradin-Szweykowska M., Peters A.J. and Lugtenburg J. The interaction of bacterioopsin with 11,14-bridged retinals. The sinthesis of 13-demethyl-11,14-imino,13-demethyl-11,14-thio-13-demethyl-11,14-etheno-11,14-imino-retinal and their binding with bacterioopsin.//J.Tetrahedron.1984.V.101.P.5537-5540.
Degani I., Fochi R. and Regondi V. The synthesis of pyrrole-2,5-dicarbaldehydes.//Synthesis.1981.N51.P.4623-4636.
Cadamuro S., Degani I., Dughera S., Fochi R., Gatti A. and Piscopo L. General methods for synthesizing 2,4-diacylpyrroles and their precursors containing one or two masked acyl groups.//J. Chem. Soc. Perkin Trans.1 1993.N22.P.273-279.
Cadamuro S., Degani I., Dughera S., Fochi R., Gatti A. and Piscopo L., A convenient general method for the synthesis of pyrrole-2,5-dicarbaldehydes.// J. Chem. Soc. Perkin Trans.1.1993.N49.P.2939-2943.
Пожарский А.Ф., Анисимова В.А., Цупак Е.Б. // Практические работы по химии гетероциклов. / Издательство Ростовского университета 1988, 151с.
Beilsteins Handbuch der Organischen Chemie. 1921 B.3 s.754.
Лазарев Н. В., “Вредные вещества в промышленности”, Ленинград, Химия, 1-3 т., 1976.
Цыгальницкий В. М., “Охрана труда и техника безопасности в микробиологических производствах”, Л-д, Химия, 1990.
Баратова А. М., Корольченко А. Я., “Пожарная опасность веществ и материалов, применяющихся в химической промышленности”, М., Химия, 1-2 т., 1990.
Общесоюзные нормы технологического проектирования “Определение категорий помещений по взрывоопасной и пожарной опасности” (ОНТП 24-86), М., ВНИИПО МВД СССР, 1986.
Бобков А. С., Блинов А. А., Николаева Т. Г., “Охрана труда при производстве и переработке полимерных материалов”, М., Химия, 1986.
Санитарные нормы проектирования промышленных предприятий СН-245-71, М., Атомиздат, 1980.
“Правила устройства электроустановок”, М., Атомиздат, 1986.
Catalog Handbook of Fine Chemicals. Aldrich 1998-1999.
Семейкин А.С., Кузьмин Н.Г. Койфман О.И. Синтез 5,15 дифенил 2,3,7,8,12,13,17,18 октаметилпорфина и его производных. // Известия вузов. Химия и химическая технология. 1988. т.31.№ 6.стр.39–44.
Paine J.B. and Dolphyn D. Pyrrole chemistry. An improved synthesis of ethyl pyrrole 2 carboxylate esters from diethyl aminomalonate. // J.Org.Chem. 1985. V.50. №26. p.5598–5604.
Синтезы органических препаратов. Сборник 2. ИЛ, 1949.
Catalog Handbook of Fine Chemicals. Aldrich 1995-1996.
Beilstein handbook of organic chemistry. 1979 v.20/5 p.64-65.
В литературном обзоре соединения пронумерованы начиная с (1).