Кислотно-каталитические процессы в нефтепереработке и в нефтехимии. Решение обратной задачи кинетики статистическими методами
Решение обратной задачи кинетики статистическими методами
В рамках рациональной стратегии структура кинетической модели задается (для каждой гипотезы) и решение обратной задачи проводится для оставшихся не отклоненных моделей. Задача сводится к оцениванию констант (параметров модели) и к сравнению качества описания эксперимента различными моделями.
Более простой случай – оценивание констант для линейных моделей (в дифференциальной или интегральной форме), например,
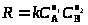
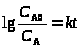
Оба уравнения – линейные функции y = bx и константа b находится методом наименьших квадратов (МНК).
В случае нелинейных моделей типа уравнений (15) или (20) решается менее строгая задача нелинейного оценивания. Параметры модели перебираются так, чтобы обеспечить минимум функционала
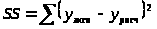 . (24)
. (24)
При этом решение является не единственным, т.е. возможно существование множества наборов констант (параметров) уравнения, одинаково хорошо описывающих эксперимент. Адекватность модели оценивается по критериям Фишера, а значимость параметров – по критерию Стьюдента.
При решении обратной задачи следует иметь ввиду и закоррелированность параметров, также не позволяющую получить единственный набор констант (кроме причин, связанных с особенностями поверхности функционала (24)). Существует проблема идентифицируемости параметров.
Пример. Рассмотрим простейшую схему каталитической реакции (схема Михаэлиса-Ментен)
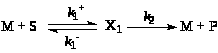 (25)
(25)
В квазистационарных условиях
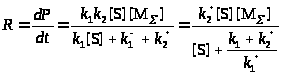 (26)
(26)
В этом простом случае
очевидно, что определяемыми
(идентифицируемыми) параметрами будут
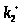 и
и
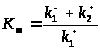 .
Этот случай неидентифицируемости
называется локальной неидентифицируемостью.
.
Этот случай неидентифицируемости
называется локальной неидентифицируемостью.
Пример. Рассмотрим случай глобальной неидентифицируемости. При анализе нестационарной последовательной реакции
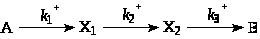
показано, что можно
определить все три константы, но решение
не является единственным. Рассчитанные
значения [А]>t>
и [B]>t>
не изменяются, если
и
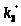 поменять местами.
поменять местами.
Таким образом, априорный анализ кинетической модели для выяснения параметров, которые могут быть оценены, является важным этапом процедуры решения обратной задачи химической кинетики.
Кислотно-каталитические процессы в нефтепереработке и в
нефтехимии
Каталитический крекинг
Каталитическим крекингом называют совокупность различных превращений углеводородов, протекающих на кислотных катализаторах (в настоящее время – гетерогенных) с образованием компонентов авиационного и моторного (автомобильного) топлива из высококипящих фракций углеводородов.
Основные реакции:
Деалкилирование (крекинг) парафинов
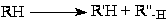
(2) Деалкилирование (крекинг) олефинов
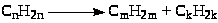 (n
= m
+ k)
(n
= m
+ k)
(3) Деалкилирование алкилароматических соединений
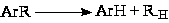
(4) Ароматизация нафтенов
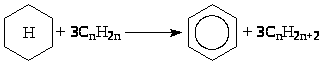
(5) Скелетная и позиционная изомеризация олефинов.
(6) Скелетная изомеризация парафинов
В современных процессах используют аморфные и кристаллические (цеолиты) алюмосиликаты Al>2>O>3> – SiO>2>. Цеолиты бывают природные и синтетические. Общая формула цеолита
M>x>D>y/2> Al>m>Si>n>O>2(m+n)>·P>H3O>, x + y = m
где M и D – одно- и двухвалентные катионы. В настоящее время только X- и Y- синтетические цеолиты используются для крекинга углеводородов. Эти цеолиты близки природным цеолитам – мордениту (X) и шабазиту (Y). При общей формуле этих цеолитов
Na>p>Al>p>Si>192–>>p>O>384>·gH>2>O
для X-цеолита P = 96 – 74, для Y-цеолита P = 74 – 48.
Кристаллическая структура цеолитов характеризуется сквозными порами одинакового диаметра (0.75 – 1.0 нм), превышающими размеры многих молекул углеводородов. При замене части ионов Na+ на ионы NH>4>+ и последующей прокалки на поверхности цеолита образуются сильные протонные центры
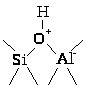
При отщеплении воды ( > 400оС) появляются апротонные кислотные центры Льюиса, локализованные на Al. Высококремнистые и термостабильные цеолиты ZSM-5 относят к очень сильным протонным кислотам (к сверхкислотам).
В основе теории механизмов реакций, протекающих в процессе каталитического крекинга, лежат представления об участии в стадиях механизмов ионов карбения и карбония и, таким образом, химия этих ионов и есть суть механизмов перечисленных выше процессов.
CH>3>+, R+ CH>5>+, RH>2>+
ионы карбения ионы карбония
На поверхности твердых кислотных катализаторов так же, как и в растворах, нет свободных ионов карбения R+. Такие частицы всегда сольватированы в растворах и переносятся на другие реагенты, освобождаясь от молекулы растворителя (как и H+).
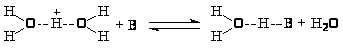
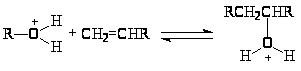
На поверхности кислотных катализаторов можно представить аналогичный процесс “сольватации” (координации) иона карбения поверхностным оксидом
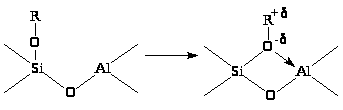
Координированный таким образом R+ аналогичен иону карбения в ионе алкоксония R>3>O+BF>4>– и может участвовать в реакциях с другими реагентами (олефинами, спиртами, аренами) аналогично реакциям алкоксониевых ионов в реакции полимеризации циклов (см. раздел 11). Ионы карбония могут находится в свободном состоянии, удерживаясь на поверхности за счет слабых водородных связей или за счет электростатических сил в виде ионных пар RH>2>+·X–.
В целом механизм процессов крекинга парафинов, олефинов, скелетной изомеризации парафинов и олефинов, реакций деалкилирования алкиларенов является цепным с кинетической точки зрения. Все эти процессы включают стадии инициирования, зарождения активных центров R+ (W>i>), стадии продолжения кинетической цепи с участием ионов карбения и карбония и стадии обрыва активных R+ (W>0>) за счет примесей в реакционной смеси. Добавки долей процента олефинов к чистому алкану резко ускоряют процесс каталитического крекинга.
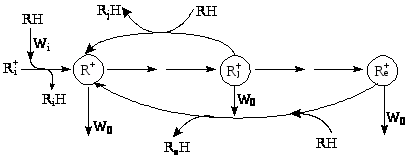
Образование кокса на поверхности алюмосиликатных катализаторов (следствие глубокого дегидрирования полимеров) является основной причиной дезактивации катализаторов крекинга, наряду с обратимым отравлением сильно адсорбирующимися примесями и необратимым отравлением металлами, содержащимися в нефтяных фракциях.
Различные функции падения активности по времени обобщены в теории, учитывающей, как важный фактор, время проведения процесса и предполагающей параллельное протекание различных процессов, ведущих к отравлению катализатора, т.е. к уменьшению количества активных центров C>S>. Уравнение (1) не включает концентраций реагентов и ядов и является, таким образом, эмпирическим уравнением, но оно было успешно применено для большого числа реакций крекинга и дегидрирования
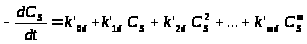 (1)
(1)
Если обозначить долю центров, сохранивших активность, = C>S>/C>S>>0>, получим
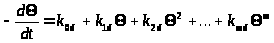 (2)
(2)
Используют и более простую модель
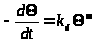 (3)
(3)
Интегрируя (3), получаем (m 1)
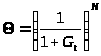 , (4)
, (4)
где G = (m – 1)·K>d>·t, N = 1/(m – 1).
Если активность катализатора пропорциональна числу активных центров
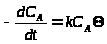 , (5)
, (5)
подставив уравнение (4) в уравнение (5), получим зависимость степени превращения X>A> сырья А как функцию концентраций, температуры и характера дезактивации активных центров.
Рассмотрим подробнее реакции скелетной изомеризации парафинов и нафтенов.
Скелетная изомеризация парафинов
Устойчивость ионов карбения растет в ряду R+>перв> < R+>втор> < R+>трет>. Поэтому процессы скелетной изомеризации парафинов термодинамически выгодны. Например, реакция (6) экзотермична
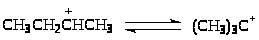
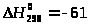 кДж/моль (6)
кДж/моль (6)
и при 25оС в равновесии отношение концентраций tBu+ / 2 – Bu+ 1010. Простейший механизм скелетной изомеризации парафинов включает 1,2-перенос метильных (алкильных) групп в первичном R+. Образование первичного R+ происходит различными путями. Главные пути – протонизация олефинов и отрыв H– подходящим акцептором (апротонным центром, ионом металла, другим ионом R+, появившимся в системе из олефинов)

1,2-Перенос алкильной группы сопровождается 1,2-гидридным сдвигом в направлении образования более устойчивого иона R+.
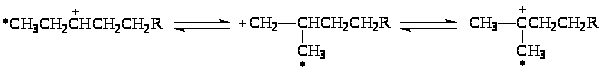
Вместе с тем, 1,2-перенос алкильной группы не объясняет факты, установленные при изучении изомеризации меченого 13С бутана. Обнаружены два изомера и две пары изотопомеров:
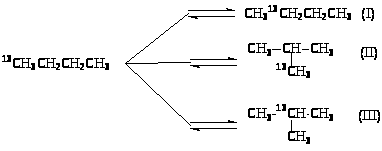
Образование изомеров I и III нельзя объяснить 1,2-переносом CH>3>-группы. Механизм этих реакций объясняют с позиций образования ионов карбония. При 1,2-сдвиге метильной группы в первичном ионе карбения образуется ион карбония (IV), который можно рассматривать как продукт протонирования циклопропанового кольца (V):
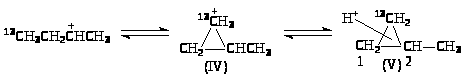
При разрыве С>1>-С>2>-связи образуются ионы карбения СH>3>13CH>2>+CHCH>3> и +CH>2>13CH>2>CH>2>CH>3>, которые приводят к продуктам I и III
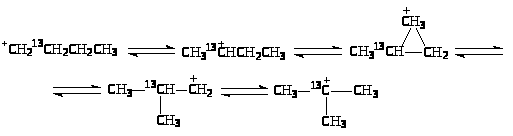
Относительные скорости взаимных превращений углеводородов в процессе скелетной изомеризации отражают более быстрое изменение положения третичного атома, чем превращения вторичного в третичный:
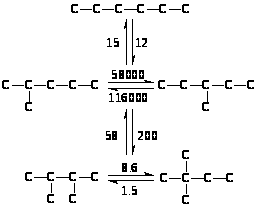
В реакциях крекинга парафинов основными продуктами являются низшие парафины, тогда как олефины (особенно С>4> и С>5>) образуют полимеры, ароматические соединения и кокс. В процессе крекинга гексадекана обнаружено 60 углеводородов от С>1> до С>10>. Условия крекинга и природа катализатора (сила кислотных центров, соотношение протонных и апротонных центров) влияет на состав продуктов.
В процессе крекинга циклопарафинов (нафтенов) образуются алифатические углеводороды и циклы меньшего размера, при этом на алюмосиликатных катализаторах обнаружено даже заметное дегидрирование циклопарафинов. Механизм дегидрирования пока-что не установлен. Механизм изомеризации циклов представляют следующей цепочкой превращений ионов карбения и карбония (или неклассических ионов карбения):
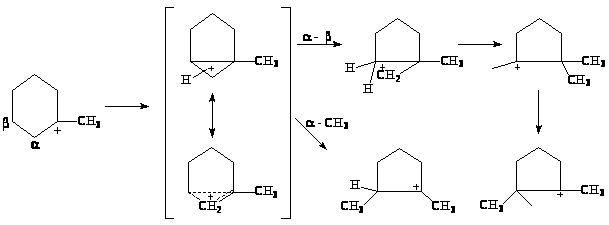
Реакции алкилирования
Реакции алкилирования – процессы обратные крекингу – занимают важное место в нефтехимии и в органическом синтезе. Реакции алкилирования используют для получения ценных компонентов моторных топлив, повышающих их октановые числа. Для получения алкилатов используют реакции алкилирования парафинов олефинами. Так, изобутан алкилируют изобутиленом и смесью изомерных бутиленов, а также пропиленом. Реакцию проводят в двухфазной системе – концентрированная H>2>SO>4> (90 – 97%) - углеводороды в присутствии ПАВ (R>4>N+X–, цетиламин, октиламин). Процесс является цепным и включает следующие типичные для кислотного катализа стадии. В концентрированных кислотах сольватация карбкатиона осуществляется молекулой кислоты
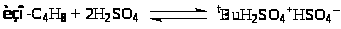 (7)
(7)
Сольватированный tBu+ взаимодействует со второй молекулой олефина с образованием димерного карбкатиона. Для упрощения изобразим схему реакций с участием свободных R+:
 (8)
(8)
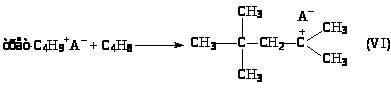 (9)
(9)
Карбкатион (VI) C>8>H>17>+ взаимодействует с изо-C>4>H>10>, отрывает от него Н– и превращается в изооктан C>8>H>18> (целевой продукт реакции). Нежелательный процесс – дальнейшее присоединение олефина с последующей полимеризацией.
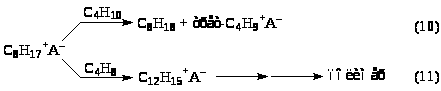
Поверхностно-активные вещества заметно увеличивают скорость реакции (10), тем самым повышая октановые числа алкилатов. Катионоподобные ПАВ повышают реакционную способность R+, уменьшают его стабилизацию кислотной фазой и делают отрыв H– от RH более эффективным.
Процессы алкилирования катализируются и другими кислотами (CF>3>SO>3>H, HF). В системе углеводород-HF-1%H>2>O процесс алкилирования протекает эффективнее и селективнее. Октановые числа алкилатов достигают 92 – 95. Предложен интересный механизм алкилирования изобутана пропиленом в присутствии HF.
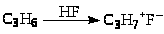

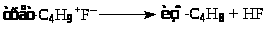
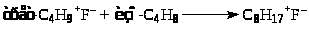
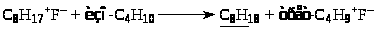
Брутто-реакция представляет собой дегидродимеризацию изобутана с переносом 2 атомов водорода на пропилен:
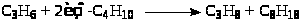 (12)
(12)
С целью заменить процессы с жидкими кислотами исследуют возможность использования цеолитов для алкилирования парафинов олефинами. Показано, что в этом случае в процесс алкилирования вступает этилен. На Na-X цеолитах при замене части Na+ ионами РЗЭ получаются достаточно высокие выходы алкилатов (80 – 100оС, Р = 2 МПа).
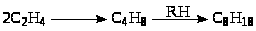
Выход C>8>-продуктов в алкилате достигает 80%.
В случае еще более сильных кислот, например, сверхкислот HF-TaF>5> с Н>0> = –18.8 алкилировать удается метан
 .
.
Среди процессов алкилирования рассмотрим две реакции, важные для органического синтеза – алкилирование бензола олефинами и алкилирование формальдегида олефинами (реакция Принса).
Алкилированием бензола получают 2 ценных полупродукта – этилбензол и изопропилбензол. Алкилирование проводят по Фриделю-Крафтсу в присутствии AlCl>3>, или на цеолитных катализаторах, которые до сих пор не используются в этом процессе в промышленности. Взаимодействуя с бензолом или с полиалкилбензолами AlCl>3>, HCl и H>2>O образуют “комплекс”, который и является катализатором реакции. Состав комплекса переменный – (ArH)>x>(H>3>O+AlCl>4>–)>y>(HCl)>z>. Вероятно, что полиалкилбензолы сольватируют H+, образуя с ним -комплекс (или -комплекс)
(ArH·H)+Al>2>Cl>7>– (VII)
который также может переносить протон на олефин с образованием R+AlCl>4>–. Реакция карбкатиона CH>3>CH>2>+ или (СН>3>)>2>СН+ с бензолом и приводит к реакции алкилирования ArH.
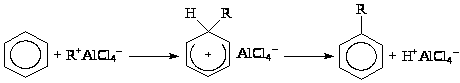
Рассматривают также возможность участия в процессе протонированного ArH (VII), однако такой механизм представляется менее вероятным,
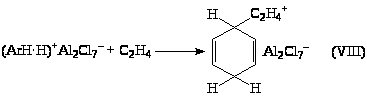
поскольку он требует ,-переноса гидрид-иона и большего числа стадий. Фирма “Monsanto” разработала процесс алкилирования, протекающий в гомогенных условиях с небольшим количеством AlCl>3> без использования жидкого катализаторного комплекса, образующего двухфазную систему. В этой технологии AlCl>3> не рециркулирует, не стабилизируется алкилбензолами, и поэтому образования полиалкилбензолов практически не происходит даже при Т > 100оС. В проточный реактор подаются этилен, бензол, AlCl>3>, HCl (и промотор). Выходящий поток, содержащий главным образом этилбензол, бензол и AlCl>3>, подают в реактор переалкилирования, куда подается диэтилбензол из узла разделения. После достижения равновесия переалкилирования
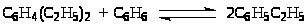
алкилат из реактора переалкилирования подается в зону испарения (удаления легколетучих и HCl), затем на промывку от AlCl>3> и в отделение ректификации.
Реакция Принса в промышленном масштабе используется в производстве изопрена. Изобутилен в растворе серной кислоты (~20%) и формальдегида превращается в диол или в 2,2-диметилметадиоксан (ДМД) в зависимости от концентрации формальдегида и условий процесса.
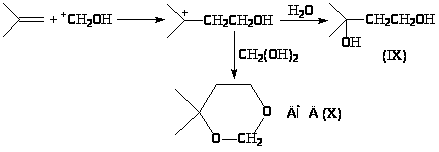
Оба продукта при дегидратации (IX) или отщеплении метиленгликоля (формальдегидгидрата) (X) образуют изопрен, однако, второй путь эффективнее – ДМД легко выделяется из реакционной среды и легко отщепляет CH>2>(OH)>2> (200оС, кислотные катализаторы). ДМД является также хорошим полярным растворителем.
Вопросы для самоконтроля:
Назовите основные реакции, протекающие в условиях каталитического крекинга.
Механизм действия кислотных катализаторов в превращениях парафинов?
Назовите общие черты механизма кислотно-каталитических превращений углеводородов.
Причины дезактивации алюмосиликатных катализаторов крекинга?
Объясните механизм образования изобутана и изотопомеров в процесс скелетной изомеризации н-бутана.
Назвать основные стадии механизма алкилирования изобутана олефинами.
Литература для углубленного изучения
Войцеховский Б.В., Корма А., Каталитический крекинг, М., Химия, 1990, 151 с.
Гейтс Б., Кетцир Дж., Шуйт Г., Химия каталитических процессов, М., Мир, 1981, с. 9 – 132.
Алкилирование. Исследования и промышленное оформление процессов, под ред. Л.Ф.Олбрайта, А.Р.Голдсби, М., Химия, 1982, с. 13 – 82, с. 114 – 130, с. 268 – 278.