Кинетика низкотемпературной радиационной постполимеризации тетрафторэтилена в стеклующихся фторорганических растворителях
КИНЕТИКА НИЗКОТЕМПЕРАТУРНОЙ РАДИАЦИОННОЙ ПОСТПОЛИМЕРИЗАЦИИ ТЕТРАФТОРЭТИЛЕНА В СТЕКЛУЮЩИХСЯ ФТОРОРГАНИЧЕСКИХ РАСТВОРИТЕЛЯХ
Способность тетрафторэтилена (ТФЭ) к полимеризации существенный образом зависит от фазового состояния, в котором находится этот мономер. При радиолизе кристаллического ТФЭ и его последующем нагревании выше температуры плавления удается получить всего лишь 1—2% полимера [1]. Низкая молекулярная подвижность мономера в кристаллической решетке, а также эффективное протекание процесса обрыва цепей в области плавления не дают возможности образоваться достаточно длинным полимерным цепям. При расстекловывании растворов мономеров резко возрастает молекулярная подвижность, обеспечивающая эффективный рост цепи при одновременном подавлении процессов обрыва, вследствие чего достаточно высока скорость конверсии [2]. Так, исследование полимеризации при расстекловывании растворов ТФЭ в матрице перфторалканов показало, что полимеризация проходит до глубоких конверсии при температурах ниже точки плавления ТФЭ [3]. Цель настоящей работы — исследование кинетики радиационной постполимеризации ТФЭ при размораживании радиолизованных стеклообразных растворов мономера в индивидуальных перфорированных соединениях.
В качестве стеклующихся растворителей использовали перфтор-2,4-диметил-3-этилпентен-2 (ФМП)
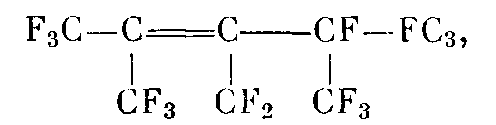
перфтор-4-метилпентен-2
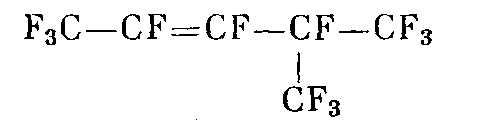
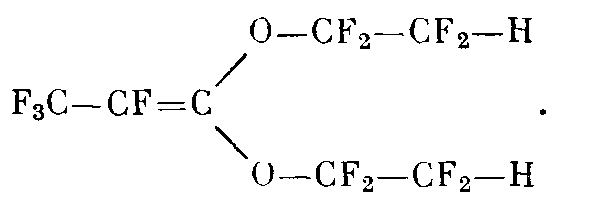
и 3-Р-гидротетрафторэтокси-6-гидро-4-оксаперфторгексец-2 (ГОГ)
Для анализа фазового состояния замороженных растворов ТФЭ и измерения скоростей полимеризации использовали калориметрическую методику [4].
Газообразный ТФЭ для очистки от ингибитора пропускали через раствор H2SO4 и колонку с активированным углем.
Навеску ФМП в стеклянной калориметрической кювете освобождали от растворенного воздуха, а затем туда же намораживали требуемое количество ТФЭ и кювету запаивали. При размораживании кюветы в ней образовывался гомогенный прозрачный раствор. Полученный раствор замораживали до 77 К и облучали лучами 60Со. Облученный образец помещали при 77 К в калориметр и следили за кинетикой тепловыделения, обусловленного полимеризацией ТФЭ в ходе размораживания. После проведения калориметрического эксперимента кювету вскрывали и после удаления растворителя и не прореагировавшего ТФЭ определяли выход полимера. Из сопоставления выхода полимера и измеренного интегрального тепловыделения была определена средняя теплота полимеризации ДЯ=(150±20) кДж/моль, т. е. практически такая же, как и определенная ранее величина для полимеризации ТФЭ в матрице перфторалканов [3]. Эту величину и использовали для расчета кинетических кривых полимеризации из калориметрических измерений.
При охлаждении со скоростью ~200 К-мин-1 ФМП полностью переходит в стеклообразное состояние. Переход из стеклообразного состояния в переохлажденную жидкость в ходе размораживания наблюдается при Г>С>^150К (рис. 1,а).
Растворы ТФЭ в ФМП (содержание ТФЭ до 23 вес.%) также полностью стеклуются, и при размораживании таких образцов на калориметрической кривой наблюдается только «ступенька» расстекловывания, отвечающая переходу стеклообразного раствора в переохлажденную жидкость. С увеличением концентрации ТФЭ в ФМП переход системы из стеклообразного состояния в переохлажденную жидкость монотонно смещается в область более низких температур (рис. 1, а, таблица). Ранее такая же тенденция наблюдалась для стеклообразных растворов ТФЭ в перфторалканах [3].
Постполимеризация ТФЭ в радиолизованных стеклообразных растворах ТФЭ и ФМП наблюдается в узком температурном интервале в области расстекловывания системы (рис. 1,6). С ростом исходной концентрации ТФЭ в растворе Т>с> монотонно смещается в область низких температур, и соответственно снижается температурная область протекания полимеризации. При этом уменьшается и интегральный выход полимера (таблица).
После достижения максимума скорость постполимеризации ТФЭ быстро падает. Это уменьшение скорости не может быть связано с выработкой мономера, поскольку его конверсия при достижении максимума скорости не превышает 10—20%. Естественно, связано это падение скорости с обрывом полимерных цепей. Обрыв растущих полимерных цепей может происходит либо из-за рекомбинации растущих макрорадикалов R>p>* с радикалами, образовавшимися при радиолизе ФМП БФМП, время жизни которых весьма велико как в твердом, так и в жидком состояниях [5]
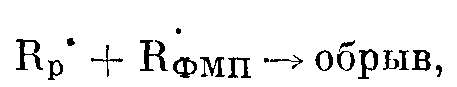
либо в реакции растущего макрорадикала ТФЭ с двойной связью ФМП с образованием малоактивного стабильного радикала, неспособного из-за стерических затруднений к дальнейшему продолжению цепи
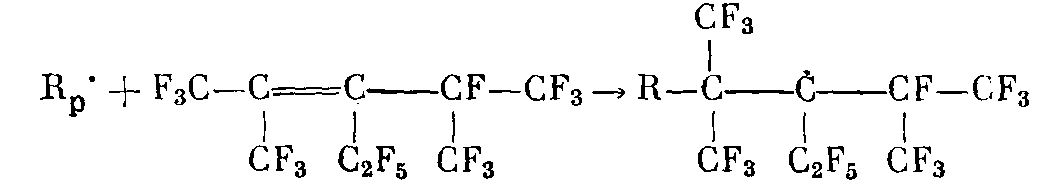
В обоих случаях должен наблюдаться линейный обрыв полимерных цепей. Тогда спад скорости постполимеризации ТФЭw во времени должен описываться уравнением
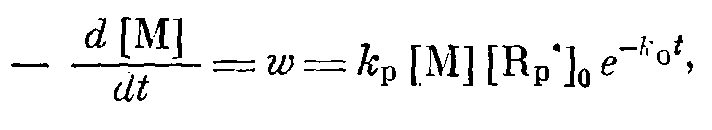
где k>f> и к>0> — соответственно константы скорости роста и обрыва полимерных цепей; [R>p>*] >0> — максимальная концентрация растущих радикалов в системе; [М] — текущая концентрация мономера. Величина [М] в условиях одного эксперимента изменяется не более чем на 10—15%, и ее убылью можно пренебречь.
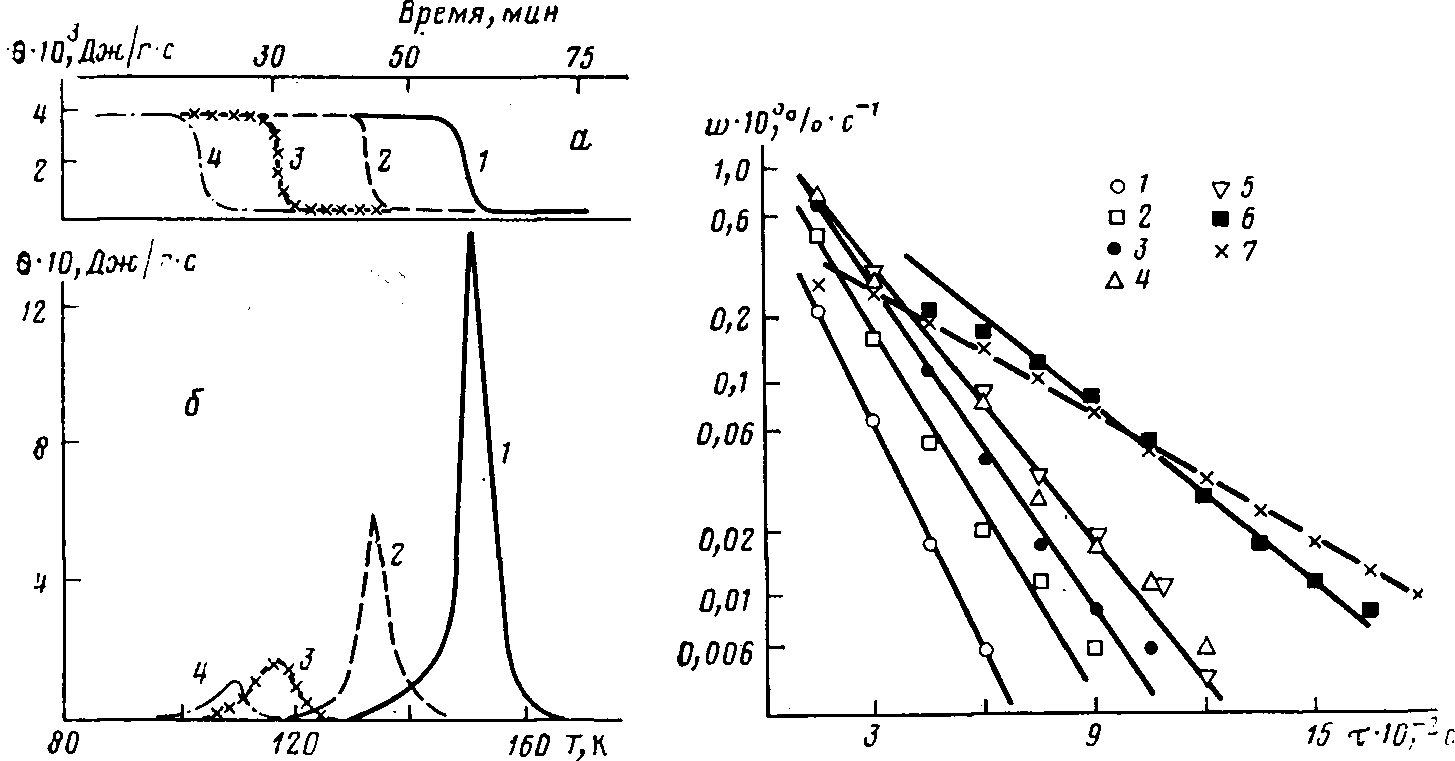
Рис. 1. Калориметрические кривые нагревания необлученных растворов ТФЭ в ФМП (а) и интегральная теплота полимеризации (б) для 1 (2), 9 (2), 17 (3), 25 вес.% ТФЭ (4)
Рис. 2. Изменение скорости полимеризации и> со временем для заключительной стадии полимеризации ТФЭ в ФМП для 1 (1), 2 (2), 4 {3), 6 (4), 9 (5), 17 (0), 25 вес.% ТФЭ (7)
Теперь, если полагать, что к>р> и к>0> в интервале температур ДГ^Ю К изменяются слабо, то спад скорости постполимеризации для каждого эксперимента, представленного на рис. 1, б, должен спрямляться в координатах lg w от t, а из наклона этих прямых можно определить величины к.
Действительно, спад скорости полимеризации во всех экспериментах (рис. 1,6) хорошо спрямляется в указанных координатах (рис. 2). Поскольку при изменении исходной концентрации ТФЭ калориметрические пики полимеризации наблюдаются в различных областях температур, то величины к>0> определены из наклона прямых на рис. 2 для разных температур (таблица). Из этих данных получена температурная зависимость к>0>, которая в аррениусовских координатах представлена на рис. 3.
Таким образом, для температурного интервала 110—155 К имеем к>а>= =0,1 ехр(-800Г) с-1.
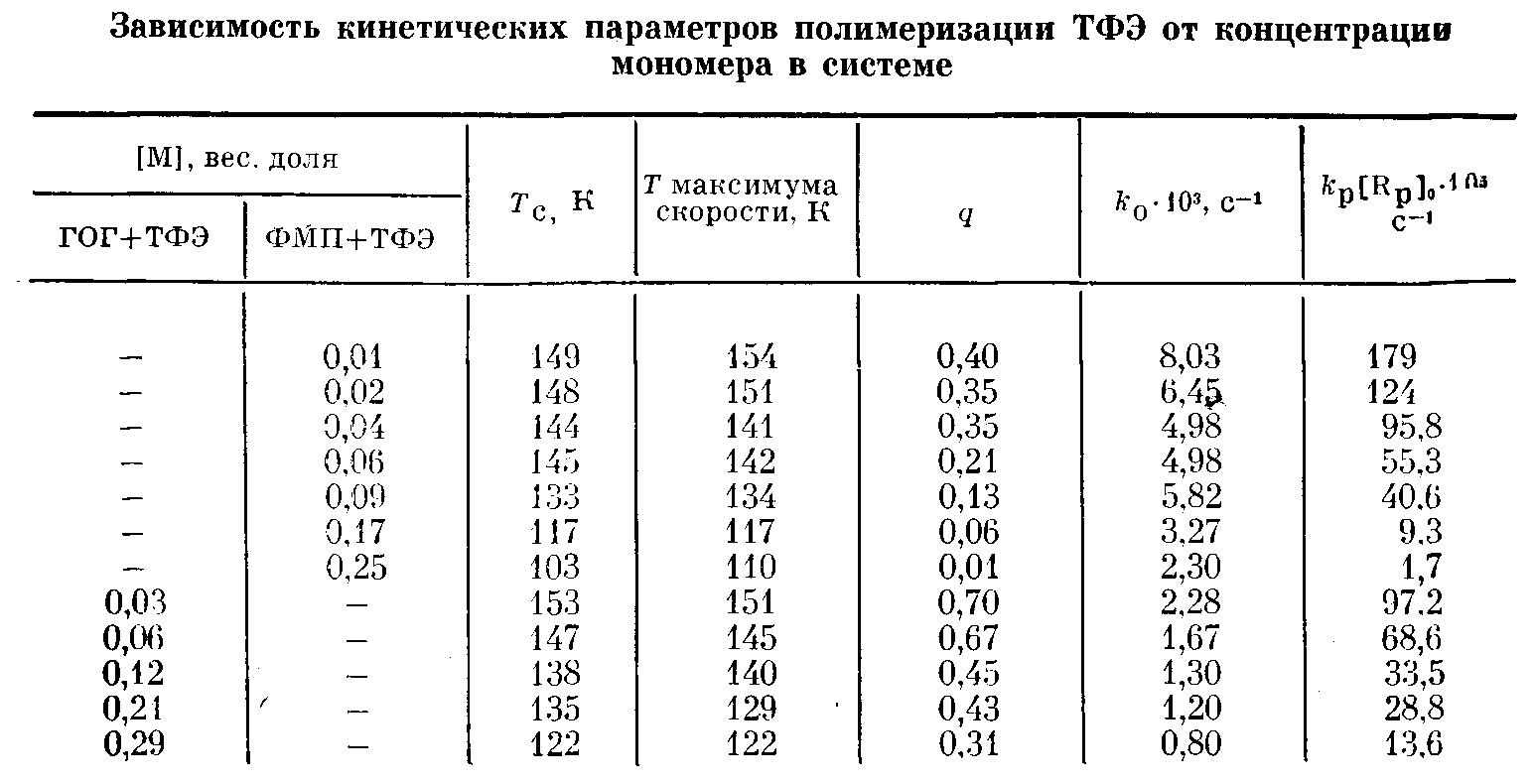
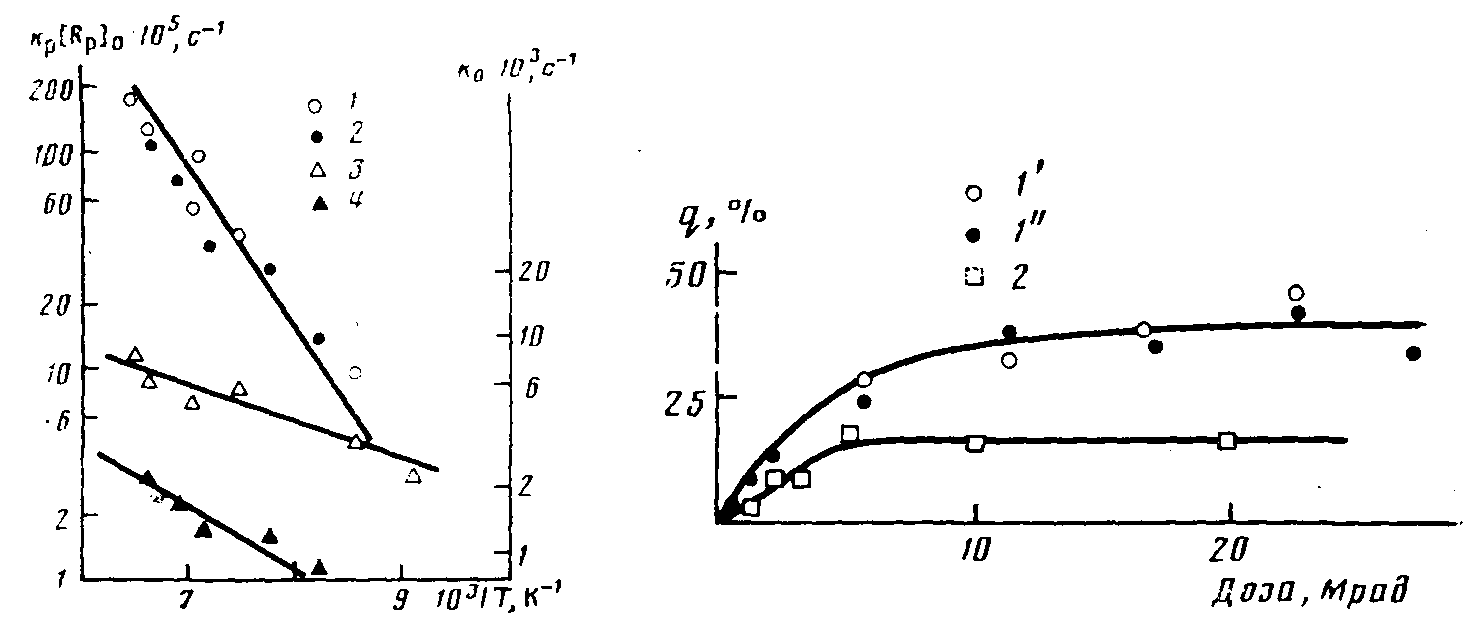
Рис. 3. Изменение константы скорости обрыва к>0> (1, 2) и константы скорости роста k>v>> >(3, 4) с температурой для процесса полимеризации ТФЭ в ФМП (1, 3) и ГОГ (2, 4)
Рис. 4. Зависимость выхода полимера от дозы предварительного облучения для 1 (1', 1") п 9 вес.% (2) раствора ТФЭ в ФМП по данным калориметрических (Г, 2) и гравиметрических (1) измерений
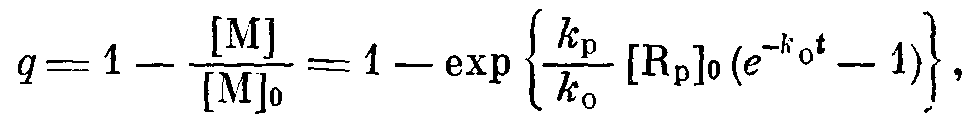
где [М] о — исходная концентрация мономера. Отсюда следует, что предельный выход полимера q (q при £>1), образовавшегося после достижения максимума скорости полимеризации, описывается следующим уравнением:
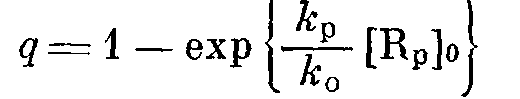
Величина g может быть легко определена из калориметрических измерений. Таким образом, для каждого из экспериментов, приведенных в таблице, известны величины к>0> и q, поэтому, пользуясь уравнением (3) можем определить величину /c>p>[R„]>0>. Поскольку условия и доза предварительного облучения во всех экспериментах не изменялись и величины радиационно-химического выхода радикалов для ТФЭ и ФМП почти не различаются, можно сделать предположение об одинаковой эффективности инициирования полимеризации и, следовательно, постоянстве величины (R>p>]o в каждом эксперименте. Поэтому изменение /e>p>[R>P>]>0> с температурой отражает лишь температурную зависимость константы скорости роста, которая на рис. 3 представлена в аррениусовских координатах. Энергия активации роста Е>р>= (14650±2100) Дж/моль. Величина предэкспоненци-ального множителя константы скорости роста может быть получена, если удается измерить величину [R>p>'] >0>- Спектры ЭПР в исследуемой температурной области представляют собой сложную суперпозицию спектров радикалов ТФЭ и ФМП, и поэтому определение концентрации растущих полимерных радикалов не представляется возможным.
Образующиеся при радиолизе ФМП (при 77 К) и следующем его размораживании стабильные радикалы (R>CT>) [5] не инициируют полимеризацию в условиях эксперимента. Действительно, после размораживания радиолизованной системы ФМПЧ-ТФЭ и проведения полимеризации (конверсия мономера 13%) по спектрам ЭПР регистрируется R>CT>. Однако при замораживании и повторном разогревании системы полимеризация не наблюдается, R>CT> не инициируют полимеризацию, как в области расстекловывания, так и при температурах выше Т>с>.
Общая концентрация радикалов, стабилизированных при радиолизе системы ТФЭ + ФМП, возрастает практически линейно с дозой облучения до —20 Мрад. Радиационно-химический выход радикалов G>R>=1,3. При размораживании радиолизованной системы ФМП + ТФЭ до 300 К, как и для чистого ФМП [5], более половины накопленных радикалов и в жидкости остается стабильным. Однако эти радикалы, как упоминалось выше, не инициируют полимеризацию. Оценки показывают, что эффективность использования накопленных при низкотемпературном радиолизе радикалов для инициирования полимеризации невысока. Так, _если предполагать, что степень полимеризации полученного полимера Р=100, то лишь 5% накопленных в ходе радиолиза при 77 К радикалов дают полимерные цепи.
С увеличением дозы предварительного облучения выход полимера в исследуемой системе монотонно возрастает и при дозах 7—10 Мрад достигает предельного значения (рис. 4). Для выяснения причины такой остановки реакции было проведено исследование влияния фотоотбеливания на процесс постполимеризации. Облучение видимым УФ-светом (Х^236 нм) стеклообразного раствора ТФЭ в ФМП при 77 К в течение 5 ч не приводит к полимеризации при размораживании, на калориметрической кривой не наблюдается тепловыделения, связанного с полимеризацией. Фотоотбеливание образца, предварительно подвергнутого f-радиолизу в тех же условиях, приводит к частичному подавлению постполимеризации, выход полимера уменьшается вдвое. Действие же УФ-света при 77 К на систему ФМП + ТФЭ, содержащую R>CT>, не приводит к образованию полимера при расстекловывании. Таким образом, совокупность полученных экспериментальных данных не дает основания предполагать, что ионные процессы играют определяющую роль в постполимеризации.
Была исследована также постполимеризация ТФЭ при расстекловывании других фторорганических растворителей. Соединение ГОГ при охлаждении полностью переходит в стеклообразное состояние (Т>с> —155 К). Растворение ТФЭ в этом соединении также приводит к смещению Т>с> в область более низких температур. Полимеризация предварительно облученных образцов протекает в области расстекловывания, как и для системы ФМП + ТФЭ, однако общий выход полимера выше (таблица). Спад скорости полимеризации для каждого из экспериментов, представленных в таблице, согласно уравнению (1), хорошо спрямляется в координатах lg w от t, что дает возможность определить величину к>0> (таблица). По уравнению (3) были определены значения c>p>[R>p>]>0>, которые также представлены в таблице. Температурные зависимости величин к>а> и [Rp] о для системы ГОГ + ТФЭ представлены на рис. 3.
Таким образом, для этой системы в температурном интервале 120— 150 К имеем с>0>=5,5ехр с-1 и к>р> [R>p>-]=180 exp{-3500/RT} с-1 Следовательно, различия процессов постполимеризации ТФЭ при расстекловывании исследованных матриц связано с различием в константах обрыва. Уменьшение k>Cl> при переходе от ФМП к ГОГ приводит, по-видимому, к увеличению средней длины образующихся полимерных цепей и, следовательно, к увеличению общего выхода полимера при одинаковой дозе предварительного облучения.
Расстекловывание перфтор-4-метилпентена-2 происходит при более низких температурах (Г>с>^112 К). Растворение ТФЭ в этой матрице приводит лишь к незначительному смещению Т>с> в область низких температур. Естественно, что полимеризация при столь низких температурах протекает с незначительными скоростями.
Таким образом, использование метода постполимеризации при расстекловывании матрицы позволило определить основные кинетические параметры низкотемпературной полимеризации ТФЭ. Для определения предэкспоненциальных множителей константы роста необходимы измерения числа растущих полимерных цепей. Подобные кинетические исследования с использованием матриц, стеклующихся при более высоких температурах, позволит в дальнейшем провести измерение кинетических параметров процесса для более широкого температурного диапазона.
ЛИТЕРАТУРА
Аллаяров С.Р., Кирюхин Д.П., Асамов М.К., Варкалов И.М. Химия высоких энергий, 1980, т. 14. № б, с. 509.
Варкалов И.М. Успехи химии, 1980, т. 49, № 2, с. 362.
Аллаяров С.Р., Кирюхин Д.П., Асамов М.К., Варкалов И. М. Высокомолек. соед. А, 1982, т. 24. № 3, с. 466.
Варкалов И.М., Кирюхин Д.П. Высокомолек. соед. А, 1980, т. 22, № 4, с. 723.
Аллаяров С.Р., Варкалов И. М., Голъданский В.И., Кирюхин Д.П. Изв. АН СССР. Сер. хим., 1983, № 6, с. 1225.