Качественный анализ компонентов
Реферат на тему:
Качественный анализ компонентов
Хроматографист, начинающий работать в области высокоэффективной жидкостной хроматографии, должен ознакомиться с основами качественного анализа. Качественный анализ применяют для идентификации известного продукта, полученного новым путем или находящегося в смеси с другими продуктами. Он необходим при выделении из сложных биологических, химических смесей различных компонентов, что особенно важно в медицине, криминалистике, экологии, для контроля за нахождением некоторых лекарств и химических продуктов и их метаболитов в биоматериалах. Знакомство с основами качественного анализа поможет избежать типичных ошибок, например, отличить примеси в образце от примесей в растворителе или проверять чистоту вещества не на одной длине волны спектрофотометра, а на разных и т.д.
Прежде чем приступить к анализу, необходимо установить, весь ли образец элюируется из колонки данной системой растворителей или нет. Чтобы быть уверенным в полном элюировании, необходимо собрать всю вытекающую из колонки жидкость, упарить растворитель, взвесить остаток и найти степень извлечения образца.
Идентификацию компонентов в ВЭЖХ можно проводить тремя способами: 1) использовать информацию об удерживании; 2) исследовать зоны, полученные при разделении в колонке жидкостного хроматографа, методами спектрального или химического анализа; 3) непосредственно подключить спектральный анализатор к колонке.
Для регистрации пиков в хроматографии используют удерживаемый объем V>R> или время удерживания t>R>. Обе величины являются характеристикой вещества в данной хроматографической системе. Так как время удерживания разделяемого вещества состоит из времени взаимодействия в колонке и времени прохождения пустых участков трубки, оно меняется от прибора к прибору. Удобно иметь вещество, не удерживаемое данной колонкой, приняв его за стандарт, время и объем удерживания которого t>0>, V>0>. Хроматографирование вещества и стандарта необходимо проводить при одних и тех же условиях (давлении и скорости потока). Поправку на мертвый объем можно учесть, используя исправленное время удерживания tn или исправленный объем удерживания V>R>, по формуле
t>R>’=t>R> — t>0>
При идентификации по данным об удерживании, известные индивидуальные вещества, которые могут присутствовать в образцах, разделяют в той же самой хроматографической системе, и для них получают значения t>R>. Сравнивая эти значения t>R >с временем удерживания неизвестного пика, можно обнаружить, что они либо совпадают, и тогда вероятно, что пики соответствуют одному и тому же веществу, либо t>R> известного вещества не соответствует t>R> неизвестной зоны. Тогда все же возможна ориентировочная оценка значений t>R> веществ, не доступных для непосредственного измерения степени их удерживания. Рассмотрим оба варианта.
В первом случае, очевидно, необходимо предварительное изучение образца для постулирования присутствия в нем конкретных веществ. При работе с простыми смесями нетрудно определить, совпадает ли степень удерживания зон образца и известных веществ, или нет, т. е. значения t>R> одинаковы или различаются. В случае сложных смесей сразу несколько веществ могут элюироваться со схожими значениями t>R>, и реально получаемые при хроматографическом разделении зоны перекрываются. В результате получение точных значений t>R> для различных зон становится невозможным. Надежность идентификации возрастает при повышении разрешающей способности, более тщательном контроле условий разделения, многократном измерении значений t>R> и усреднении найденных величин. При этом хроматографическое разделение известного и неизвестного веществ должно чередоваться. При разделении сложных смесей значение t>R> вещества может изменяться под влиянием матрицы самого образца. Такое воздействие возможно в начале хроматограммы и при перекрывании пиков; возможно также затягивание зон, о чем уже упоминалось.
В подобных случаях следует добавить стандарт к образцу в соотношении 1:1. Если вещества идентичны, значение t>R> исходного вещества не изменится, и на хроматограмме получают только один пик. Если имеется прибор с циклической системой хроматографирования, то для надежности идентификации желательно смесь пропускать через колонку несколько раз.
Сведения о степени удерживания можно найти и в литературе, однако ценность этой информации ограничена. Так как колонки даже одной партии дают плохую воспроизводимость, литературные значения не всегда соответствуют истинному значению t>R> на данной колонке. Для адсорбционной хроматографии возможно, однако, предсказание t>R> на основании литературных данных. Другая трудность, связанная с использованием литературных значений t>R>,—сложность их поиска в специальной литературе, хотя библиографические обзоры, публикуемые в Jornal of chromatography, имеют обновляемый указатель по типам веществ.
Во втором случае, когда времена удерживания известных соединений и зон образца не совпадают, имеется возможность предсказать время удерживания неизвестного компонента. Вполне надежны предсказания относительного удерживания на основании данных о структуре в пространственно-эксклюзионной хроматографии. Менее точны они в адсорбционной, распределительной хроматографии и особенно при работе на химически привязанной фазе. Для ионной и ион-парной хроматографии веществ с известной р Ка возможны лишь приблизительные определения значений t>R>. Всегда легче предсказать относительное удерживание или значение α, чем абсолютные значения k’. Относительные значения t>R> легче оценить для родственных соединений или производных, например замещенных алкилкарбоновых кислот или производных бензола.
При изократическом разделении гомологов или олигомеров иногда наблюдается следующая закономерность:
lgk'=A+Bn,
где A и В — константы для ряда выбранных образцов и для данной хроматографической системы (на одной и той же колонке, с такой же подвижной и неподвижной фазами); n—число одинаковых структурных единиц в молекуле образца.
В градиентном режиме при линейном изменении силы подвижной фазы уравнение приводится к виду
t>R> =A'+B'n,
т. е. значения t>R> линейно изменяются с изменением n.
Введение в молекулу образца функциональной группы j будет приводить к изменению k' в первом уравнении на некоторый постоянный коэффициент αj в данной хроматографической системе. Можно получить групповые константы αj для различных замещающих групп j, значения которых будут возрастать при увеличении полярности функциональных групп, во всех видах хроматографии, кроме обращенно-фазной, где значения констант будут уменьшаться с увеличением полярности.
Некоторые групповые константы а для различных замещающих групп j приведены в табл. 1.
В адсорбционной хроматографии первое уравнение не всегда применимо, так как оно справедливо при условии, что все изомеры имеют одно и то же значение k', что не всегда соблюдается. Можно, однако, построить график зависимости lgk/ одних и тех же соединений на одной колонке относительно lgk' тех же соединений, но на другой колонке или относительно соответствующих характеристик в тонкослойной хроматографии, например, lg[(1—Rf)/Rf], и при этом обычно получают линейную зависимость.
При сопоставлении данных об удерживании веществ можно использовать значения коэффициента емкости k', так как на него в отличие от t>R> не влияют скорость подвижной фазы и геометрические особенности колонки.
Разделение на химически привязанной фазе аналогично разделению по методу распределительной хроматографии с аналогичными фазами, и поэтому данные по экстракции при равновесном состоянии могут быть использованы для предсказания времени удерживания.
Таблица 1. Групповые константы αj для различных замещающих групп j в разных жидкостных хроматографических системах (значения указаны для замещения в ароматическом кольце)
|
Замещающая Группа |
Адсорбционное разделение на силикогели |
Распределительное разделение на обащенной фазе вода – н-октанол |
Разделение на обращенной химически привязанной фазе с отношением метанол – вода (1:1) |
|
-Br -Cl -F -CH3 ^-CH3 -O-CH3 -NO2 -CN -CHO -CO2CH3 -COCH3 -OH -NH3 -COOH -CONH3 |
0.5 0.15 0.7 0.8 1.7 2.1 2.3 3.7 4.9 5.2 22 22 55 200 450 |
7.2 5.1 1.4 3.6 4.1 1.0 0.5 0.3 1.0 0.3 0.2 0.06 0.50 |
2 1.9 0.9 0.7 0.5 0.5 1.1 0.6 0.3 0.3 0.1 |
В ионообменной хроматографии на степень удерживания влияют три фактора: степень ионизации кислот и оснований, заряд ионизированной молекулы и способность вещества из водной подвижной фазы, используемой в ионообменной хроматографии, мигрировать в органическую фазу. Последнее зависит от молекулярной массы соединения и его гидрофобности. Следовательно, более сильные кислоты или основания сильнее удерживаются при анионообменном или катионообменном разделении. При снижении рКα отдельной кислоты, входящей в образец, удерживание возрастает при разделении ряда кислот за счет анионного обмена, а при увеличении рКα увеличивается удерживание оснований при их разделении за счет катионного обмена.
Таким образом, совпадение значений времени удерживания известного вещества с наблюдаемым дает возможность предположить их идентичность. Достоверность идентификации возрастает, если проводить сравнение хроматограмм известного вещества и неизвестного компонента в различных условиях. Если вещества в адсорбционной и обращенно-фазной или ионнообменной и эксклюзионной хроматографии ведут себя одинаково, надежность идентификации возрастает. Если достоверность идентификации при равенстве относительного удерживания составляет 90%, то при изучении поведения этих же веществ в условиях существенно отличающихся достоверность идентификации составляет уже 99%.
Ценной характеристикой вещества, применяемой при идентификации, является отношение сигналов, полученных для данного вещества на двух разных детекторах. Анализируемое вещество после выхода из колонки проходит сначала через первый детектор, затем через второй, а сигналы, поступающие с детекторов, регистрируются одновременно при помощи многоперьевого самописца или на двух самописцах. Обычно применяют последовательное соединение ультрафиолетового детектора (более чувствительного, но селективного) с рефрактометром, или ультрафиолетового с детектором по флуоресценции, или двух ультрафиолетовых детекторов, работающих на разных длинах волн. Относительный отклик, т. е. отношение сигнала рефрактометра к сигналу фотометра, является характеристикой вещества при условии, что оба детектора работают в своем линейном диапазоне; это проверяется введением различных количеств одного и того же вещества. Качественную информацию можно получить, работая на фотометрических детекторах, снабженных устройством для остановки потока (Stop flow) и позволяющих регистрировать спектр выходящего из колонки пика, пока он находится в проточной кювете, сравнивая его со спектром известного соединения.
Значительный интерес при идентификации представляют современные, пока еще дорогие, спектрофотометры с диодной решеткой.
Совершенно неизвестное вещество невозможно идентифицировать только с помощью высокоэффективной жидкостной хроматографии, необходимы и другие методы.
ИДЕНТИФИКАЦИЯ ВЕЩЕСТВ НЕХРОМАТОГРАФИЧЕСКИМИ МЕТОДАМИ
В некоторых случаях идентификация неизвестного вещества может быть обеспечена сбором фракции, соответствующей пику хроматографического разделения, и последующим анализом этой фракции физическими или химическими методами. При этом подвижная и неподвижная хроматографические фазы должны быть очищенными, чтобы фон от фазы был сведен к минимуму, они не должны вступать в химическую реакцию с растворенным веществом, должны быть совместимыми-с хроматографической системой, используемой для разделения и обнаружения пика. Неподвижная фаза не должна выноситься из колонки. Кроме того, обе фазы не должны мешать идентификации вспомогательными методами и быть летучими, чтобы их можно было легко удалить выпариванием, фракции обычно собирают вручную, хотя возможно применение коллектора фракций. Для обеспечения чистоты, соответствующей пику собираемой фракции, внутренний объем трубки между детектором и выходом канала для сбора фракций должен быть минимальным. Этот объем должен быть измерен и внесены поправки на задержку между регистрацией пика детектором и фактическим выходом пика из канала для сбора фракций. Фракции удобно собирать в чистые, сухие, защищенные от попадания света сосуды с навинчивающимися крышками и тефлоновыми прокладками во избежание загрязнений. Возможен барботаж этих фракций чистым азотом или гелием. Растворители удаляют из образца выпариванием, продувкой газом, нагреванием ИК-лампой. Воду и смеси органических растворителей с водой удаляют выпариванием или лиофильной сушкой. Летучие буферные соединения удаляют при повышенных температурах.
Масс-спектрометрия — наиболее надежный и полный метод определения структуры вещества, молекулярной массы, его химической формулы. Этот высокочувствительный метод позволяет работать всего с 0,005 мкг вещества и установить как фрагментарный состав молекулы, так и ее элементное строение.
Рассмотрение идентификации неизвестных веществ с помощью масс-спектрометрии не входит в задачу данного издания. Следует отметить, однако, что большинство неионных, даже относительно малолетучих веществ удается проанализировать, используя обычно применяемый в масс-спектрометрии метод возбуждения электронным ударом. Нелетучие или ионные соединения определяют при использовании методов химической ионизации, полевой десорбции и других специальных приемов.
Образец, предварительно упаренный до 1—2 капель (объем 25—50 мкл), переносят микрошприцем во входной зонд масс-спектрометра, медленно выпаривают и анализируют. При этом необходимо выполнять холостые опыты, отбирая аналогичную фракцию до выхода интересующего нас пика, упаривая ее и вводя в масс-спектрограф, чтобы убедиться в отсутствии фоновых примесей, которые могут дать значительные пики и мешать правильной идентификации.
Для спектрального анализа в инфракрасной области требуется относительно небольшая проба — 5 мкг в твердом виде и 50 мкг в растворе. Чувствительность повышается при использовании методов, основанных на преобразовании Фурье, при этом достаточно всего 0,05 мкг пробы. Этот метод анализа прекрасно дополняет данные, полученные на масс-спектрометре, и дает информацию о функциональных группах, а иногда и о структуре вещества. Имеется несколько приемов, позволяющих анализировать разделенные на хроматографе вещества с помощью ИК-спектрофотометра. Наиболее простым является концентрированно на таблетке КВч. Собранную с хроматографа и упаренную до 1—2 капель фракцию наносят микрошприцем на 5—10 мг порошка бромида калия, причем каждую новую порцию раствора выпаривают на таблетке, пропуская сухой ток инертного газа.
Рис.1. Сосуд для концентрирования образца на микротаблетке КВг для спектрального анализа:
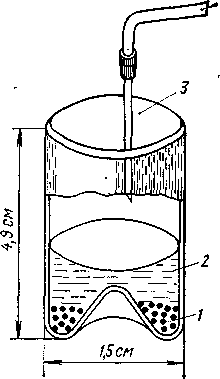
1 — КВг; 2 — собранная фракция; 3 — игла для подкожных инъекций; 4 — подача сухого азота
Другой путь состоит в выпаривании всей собранной фракции в сосуде с выпуклым дном (рис. 1), на котором находится порошок бромида калия. Растворитель полностью удаляют, а порошок бромида калия перемешивают и спрессовывают в микротаблетку. Таблетку из бромида калия можно непосредственно перенести в зону масс-спектрометр или в ИК-спектрофотометр. Можно вводить концентрированную фракцию в микрокювету спектрофотометра и сканировать, используя компенсирующий растворитель. Метод, основанный на преобразовании Фурье, дает довольно высокую чувствительность.
Применяют метод анализа в ИК-области с использованием комбинационного рассеяния. Растворитель, содержащий вещество, выпаривают на специальной пластинке, на которой остается тонкая пленка, после чего снимают спектральную характеристику. И здесь для повышения чувствительности желательно иметь прибор с преобразователем Фурье.
Для идентификации хроматографических пиков можно использовать спектры, определяющие структуры, содержащие 1Н, 11В, 13С, 19F, 31Р. Хотя для спектров ЯМР требуется большое количество образца (до 1000 мкг), при применении методов, основанных на преобразовании Фурье, значительно повышается чувствительность, так как многократное сканирование улучшает отношение сигнала к шуму и позволяет работать с малыми (до 5 мкг) количествами образца. Растворитель удаляют из хроматографической фракции полностью и заменяют его подходящим для ЯМР растворителем.
Из других методов идентификации следует отметить элементный анализ, требующий от 200 до 1000 мкг вещества, полярографию (0,01 мкг вещества), спектрофлуориметрию-,(0,05 мкг вещества) и, наконец, спектральный анализ в УФ- и видимой областях (0,1 мкг).
Простым, надежным и удобным способом определения функциональных групп являются цветные селективные реакции, для которых требуется всего 0,1 мкг вещества. Совпадение времени удерживания и соответствующая цветная реакция являются надежным подтверждением правильности структуры. Иногда достаточно растворитель, применяемый в жидкостной хроматографии, лишь частично упарить, а в некоторых случаях его следует полностью заменить подходящим растворителем. В табл. 2 приведены примеры цветных реакций для опознавания функциональных групп.
Таблица 2. Качественные реакции определения функциональных групп
|
Тип вещества |
Реактив |
Приобретенный цвет Предел обнаружения, мкг |
Обнаруживаемые вещества |
|
|
Спирты Альдегиды Кетоны |
Бихромат калия, азотная кислота Нитрат церия 2,4-Динитрофенол Реактив Шиффа 2,4-Динитрофенол |
Голубой Янтарный Желтый Розовый Желтый осадок |
20 100 20 50 20 |
C1-C8 C1-C8 C1-C6 C1-C6 C^-C8 |
|
Эфиры |
Гидроксамат железа |
Красный |
40 |
C1-C5 |
|
Меркаптаны |
Нитропруссид натрия Изатин Ацетат свинца Нитропруссид натрия |
Красный Зеленый Желтый осадок Красный |
50 100 100 50 |
C1-C9 C1-C9 C1-C9 C2-C12 |
|
Сульфиды |
||||
|
Дисульфиды |
Нитропруссид натрия Изатин Реактив Гинзберга Нитропруссид натрия Гидроксамат железа — пропиленгликоль Формальдегид, серная кислота |
Красный Зеленый Оранжевый Красный, голубой Красный |
50 100 100 50 40 |
C2-C6 C2-C6 C1-C4 C1-C4 C2-C5 |
|
Амины Нитрилы |
||||
|
Ароматические соединения |
Бордовый |
20 |
C6H^-C6H4-C4 |
|
|
Алифатические непредельные соединения |
То же |
Бордовый |
40 |
C2-C8 |
|
Галогеналкилы |
Спиртовый раствор AgNOa |
Белый осадок |
20 |
C1-C5 |
СПЕКТРАЛЬНЫЙ АНАЛИЗ НЕПОСРЕДСТВЕННО В ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЕ
Разработаны комплексные автоматизированные системы, позволяющие вводить вещество после разделения в колонке хроматографа непосредственно в спектрофотометр, но подобное оборудование является довольно дорогостоящим, требует значительной подготовки оператора и имеет ряд ограничений. Для многих задач применение таких систем нецелесообразно, а иногда даже непригодно.
Как и в случае газовой хроматографии существует несколько систем сопряжения с масс-спектрометром. Эти системы, естественно, должны обеспечивать эффективный перенос пика растворенного вещества, иметь высокую чувствительность при степени извлечения образца более 30% и давать возможность выбора нужного вида жидкостной хроматографии и режима работы масс-спектрометра. Такая система должна гарантировать быстрый, надежный анализ при минимальных требованиях к подготовке оператора. Необходимо также обеспечить возможность быстрого и полного удаления растворителя, примененного в качестве подвижной фазы; наконец, перенос пика должен быть воспроизводимым.
Возможно подключение к жидкостному хроматографу ИК-спектрометра с преобразователем Фурье. При этом химическая структура веществ, соответствующих пикам на хроматограмме, может быть идентифицирована с помощью данных, записанных в библиотеку спектральной информации системы.
Литература
Энгельгардт X. Жидкостная хроматография при высоких давлениях: Пер. с англ. М., Мир, 1980. 245 с.
Жидкостная колоночная хроматография/Под ред. 3. Дейла, К. Мащека, Я. Янака. Пер. с англ, (в 3-х томах). М., Мир, 1978.
Остерман Л. А. Хроматография белков и нуклеиновых кислот. М., Наука, 1985. 536 с. 9 Berendsen G. E., DeGalan R./l. Chromatogr., 1980, v. 196, No. 1, p. 21—37.
4. Cooke N.H. C., Olsen /(./J. Chromatogr. Sci., 1980, v. 18, p. 1—12.
5. Scottt C. D., Lee N. E./J. Chromatogr., 1973, v. 83, p. 383—393.
6. Kabra P. M., Marion L. J. Liquid Chromatography in Clinical Analysis. Humana Press, Cliftond, 1981. 466 p.
7. Edelson E. H., e. a./J. Chromatogr., 1979, v. 174, p. 409—419.