Инновационный путь развития технологии создания новых лекарственных средств
ГОСУДАРСТВЕННОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
КАЗАНСКИЙ ГОСУДАРСТВЕННЫЙ МЕДИЦИНСКИЙ УНИВЕРСИТЕТ МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ И СОЦИАЛЬНОГО
РАЗВИТИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
Биотехнологический факультет
Кафедра Общей Химии
Курсовая работа
по дисциплине «Химическая технология БАВ»
Инновационный путь развития технологии создания новых лекарственных средств
Выполнил студент 1 группы 4 курса Калинин Д.С
Проверил к.т.н., доцент Артемьева К.В
Курск-2010
Содержание
Введение
1. Методы молекулярного моделирования в основе направленного поиска лекарственных средств
1.1 Создание компьютерной модели молекулы
1.2 Описание модели квантово-химическими расчетами
1.3 Определение биологической активности по модели
2. Применение компьютерного моделирования в современной практике
3. Примеры применения молекулярного моделирования
3.1 Определение механизма взаимодействия медиатора и рецептора с ис пользованием молекулярного моделирования на примере ГАМК
3.2 Определение биологической активности при помощи программы PASS
3.3 Поиск физиологически активных аналогов ССК-4
4. Анализ примеров
Заключение
Список использованных источников
Введение
После распада СССР и государственного экономического кризиса 1998 года химико-фармацевтическая промышленность пришла в упадок. На данный момент объем продаж импортных готовых лекарственных средств составляет 75% по стоимости и 35% по объему. По мнению Российской Ассоциации Фармацевтического Маркетинга для возрождения химико-фармацевтической промышленности требуется следующее:
введение новых международных стандартов, например GMP;
внедрение новых инновационных и эффективных технологий;
разработка новых эффективных лекарственных препаратов;
привлечение инвесторов;
повышение квалификации кадров. [1]
В 2004 году Министерство здравоохранения и социального развития РФ подтвердило актуальность проблемы отсутствия новых отечественных лекарственных субстанций на рынке. Было предложено решение проблемы при помощи использования новой технологии биоскрининга активных веществ на базе «Исследовательского института химического разнообразия» (г.Химки Московская область)[2].
Биоскрининг — это набор одинаковых тестов, которые проводят c разного рода биологическими объектами, с целью выявить среди них несколько особенных экземпляров, обладающих выделяющимися свойствами. За счет использования информационных технологий и вычислительной техники сильно сокращается время биоскрининга. Примером таких информационных технологий является компьютерное моделирование новых лекарственных препаратов. В данной курсовой работе рассмотрена возможность разработки технологической схемы направленного компьютерного поиска новых лекарственных средств.
1. Методы молекулярного моделирования в основе направленного поиска лекарственных средств
1.1 Создание компьютерной модели молекулы
Вначале создается компьютерная модель объекта, а также применяется компьютерное моделирование для формирования молекул на месте проведения исследования. Модель может быть как двухмерной, так и трехмерной.
Для создания двухмерной модели используется теория графов. Граф - это абстрактная структура, которая содержит узлы, соединенные ребрами. В молекулярных графах узлами соответствуют атомы, а ребра - связям между атомами. Часто в молекулярных графах опущены атомы водорода. Узлы и ребра могут иметь свойства связанные с ними. Например, для узлов это может быть атомное число или тип атома, а для ребер число связей. Эти свойства могут быть полезны при проведении операций над молекулярным графом. Графы дают только представление о том, в каком порядке связаны между собой атомы в молекуле. Подграфы – это подмножество узлов и ребер в графе, например, бензол является подграфом молекулярного графа аспирина. Таким образом, данный граф может выполнен различными способами и не соответствовать стандартному изображению химической структуры. Изображение в виде «дерева» - это особый вид графа, в котором есть только один единственный путь подключения каждой пары вершины, то есть, нет циклов и ароматических колец.
Также необходимо иметь средства для распространения информации о молекулярных графах от компьютера к компьютеру. Это достигается различными способами. Наиболее общий способ это использование таблиц соединения. Альтернативный путь представления и распространения молекулярного графа через использование линейного описания. Недавно нашло широкое признание линейное представление - упрощенная молекулярная внутренняя линейная вводная спецификация (Simplified Molecular Input Line Entry Specification (SMILES)). Одна из причин широкого использования SMILES заключается в том, что всего несколько правил необходимо для написания и понимания большинства SMILES строк.
Такая интерпретация молекул может быть названа «естественным языком» органической химии. Тем не менее, они лишь указывают на атомы и порядок их связывания. Стерические и электронные свойства могут зависеть от того, в каком положении в пространстве находятся трехмерная структура или конформация. Таким образом, это придает особый интерес к разработке алгоритмов и систем баз данных, содержащих информацию о трехмерной структуре и конформации.
Но использование трехмерной структуры связано с рядом проблем. Большинство молекул, представляющих интерес, может иметь более одной низкоэнергетической конформации и во многих случаях количество доступных структур очень велико. Поэтому необходимы эффективные алгоритмы, принимающие во внимание конформационную гибкость. Истинное представление о молекулярных свойствах и характеристиках могут быть получены очень сложными вычислительными моделями, разработанными на основе квантовой механики и молекулярной симуляции. Эти вычислительные модели не разработаны для работы с большим числом молекул и поэтому необходима разработка более эффективных вычислительных методов для представления ключевых характеристик молекулярных конформаций.
Данные о трехмерной структуре вещества получают из экспериментальных данных ядерно-магнитного резонанса или рентген - кристаллографии. После обработки экспериментальных данных представление о трехмерной структуре записываются в специальную базу данных. К примеру Кембриджская структурная база (CSD) данных содержит данные о структуре, полученных методом рентген - кристаллографии, более чем 400,000 органических и органометаллических веществ. Протеиновая база данных содержит информацию о более чем 44,000 структурах протеинов, протеин-лигандных комплексов, нуклеиновых кислот, гидрокарбонатных структурах, полученных в результате рентген-кристаллографии и ядерно-магнитного резонанса. Обе базы данных широко используются и постоянно обновляются.
Главное применение в фармации баз данных трехмерных структур обнаружение структур, пространственная конфигурация которых позволяет взаимодействовать с конкретной биологической мишенью. Обычно это выражают при помощи трехмерных фармакоров. Под трехмерным фармакором обычно понимают набор функциональных групп с их специфичной пространственной ориентацией[3].
1.2 Описание модели квантово-химическими расчетами
В разумности модели молекулы, используемой для квантово-химических построений, согласно которой анализу подлежит система ядер и электронов и ее поведение описывается уравнениями квантовой теории, сомнений нет. Вся совокупность экспериментальных данных, полученных разными методами, не противоречит этой модели. Трудности получения химически значимых результатов на ее основе связаны с тем, что она слишком общая и всеобъемлющая, так что численное решение уравнений представляет крайне сложную задачу. Приходится делать немалое число шагов на пути создания практичных алгоритмов расчетов свойств молекул, межмолекулярных комплексов и твердых тел.
Построение поверхностей потенциальной энергии (ППЭ) представляет важнейшую составную часть компьютерного эксперимента в химии, поскольку информация, содержащаяся в детальной картине этих поверхностей для молекулярной системы, поистине стоит тех серьезных затрат, которые неизбежны даже с применением мощной вычислительной техники.
Прежде всего на поверхностях потенциальной энергии находят стационарные точки, то есть координаты минимумов, максимумов, седловых точек. Для того чтобы можно было говорить о существовании стабильной молекулы или молекулярного комплекса, на потенциальной поверхности основного электронного состояния должен быть минимум, энергия которого меньше энергии любой совокупности фрагментов, на которые можно разбить молекулу. Если этих минимумов несколько, то для молекулы возможно несколько изомеров. Координаты ядер, отвечающие точкам минимумов, определяют равновесные геометрические конфигурации, а энергии по отношению к соответствующим пределам диссоциации на составные части — энергии связи химической системы. Знание положений и энергий седловых точек необходимо для оценок энергий активации при рассмотрении элементарных химических реакций. Наличие минимумов с энергией выше предела диссоциации указывает на возможность образования интермедиатов в системе реагирующих молекул. Рассчитывая разности электронных энергий различных электронных состояний для тех геометрических конфигураций ядер, которые отвечают точкам минимумов, можно интерпретировать или предсказывать электронные спектры молекул.
После аппроксимации фрагментов потенциальных поверхностей в окрестностях точек минимумов можно переходить к рассмотрению движений систем ядер молекулы и прогнозировать или интерпретировать колебательно-вращательные спектры. Зная набор электронных колебательно-вращательных энергий молекулы можно с помощью формул статистической термодинамики вычислять любые термодинамические функции данного вещества. Если рассматривается молекулярная система, в которой возможно перераспределение частиц, то есть химическая реакция, то рассчитывается сечение потенциальной поверхности вдоль пути наименьшей энергии, связывающего реагенты и продукты, и затем оценивается константа скорости элементарной химической реакции. Описанная программа действий реализует схему расчетов свойств веществ без привлечения каких-либо эмпирических данных, которая представлена на рисунке 1.
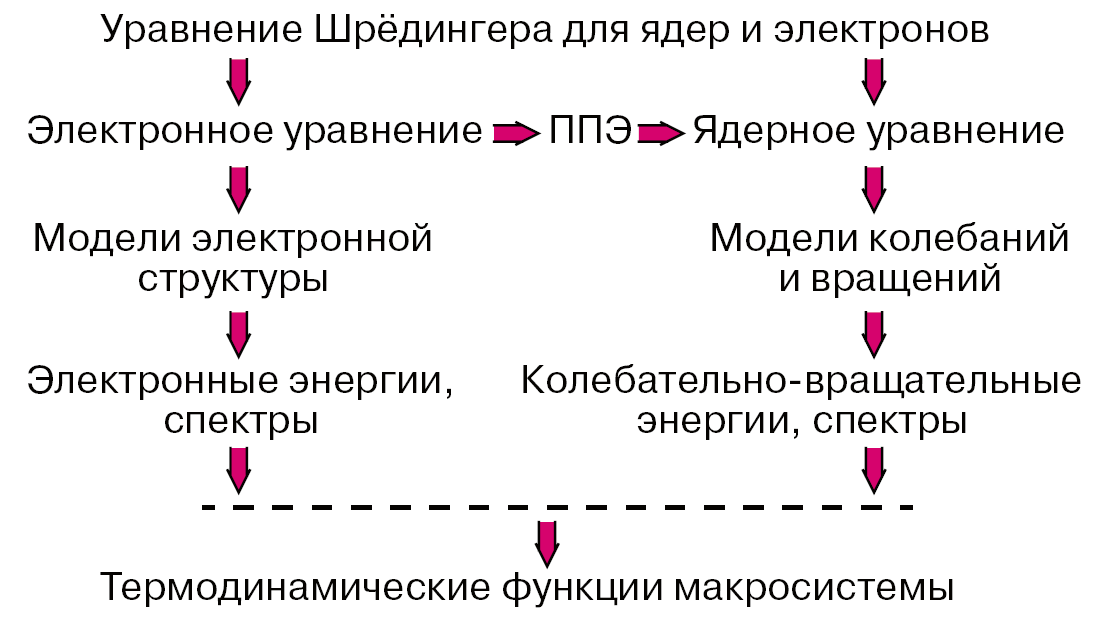
Рисунок 1 – Реализация программы расчетов свойств веществ из первых принципов
При этом результатом моделирования, которое немыслимо без компьютеров, является теоретический прогноз. Естественно, на любом промежуточном этапе этой схемы можно привлекать доступную экспериментальную информацию и вносить в компьютерное моделирование эмпирические элементы: при правильно сформулированной задаче ценность предсказаний не уменьшается, а становится более надежной. Расчеты геометрического строения и колебательных спектров молекул активно проводятся экспериментаторами, подтверждая результаты измерений квантово-химическим моделированием [4,5,6].
Молекулярная механика представляет собой совокупность методов априорного определения геометрического строения и энергии молекул на основе модели, в которой электроны системы явно не рассматриваются. Поверхность потенциальной энергии, которая в квантово-химических моделях подлежит прямому расчету, здесь аппроксимируется определенными эмпирическими функциями разной степени сложности, представляющими собой, например, суммы парных потенциалов взаимодействия атомов. Эти потенциальные функции, определяющие так называемое силовое поле молекулы, содержат некоторые параметры, численное значение которых выбирается оптимальным образом так, чтобы получить согласие рассчитанных и экспериментальных характеристик молекулы. В простейшем случае параметрами являются равновесные межъядерные расстояния и валентные углы, а также силовые постоянные, то есть коэффициенты жесткости упругих сил, связывающих пары атомов. Метод основан на допущении возможности переноса этих параметров из одной молекулы в другую, так что численные значения параметров, подобранные для некоторых простых молекул, используются далее при прогнозировании свойств других более сложных соединений.
Простейшие модели молекулярной механики учитывают растяжения связей, деформацию валентных и двугранных углов, взаимодействие валентно несвязанных атомов, называемое также Ван-дер-Ваальсовым взаимодействием, электростатические вклады и т.д.:
 ,
(1)
,
(1)
где U>раст> – энергия растяжения связей;
U>деф> – энергия деформацию валентных углов;
U>торс> – энергия деформацию двугранных углов;
U>вдв> – энергия Ван-дер-Ваальсового взаимодействия;
U>эл-стат >– энергия электростатических вкладов.
Для каждого слагаемого записывается определенное аналитическое выражение и параметры соответствующих функций подгоняются по каким-либо свойствам базовых молекул. Например, для описания потенциальной функции предельных углеводородов при не очень высоких требованиях к точности расчета достаточно около десяти параметров.
Сумма всех перечисленных вкладов определяет энергию U молекулы как функцию геометрической конфигурации ядер, и для нахождения равновесной геометрической конфигурации исследуемой молекулы необходимо определить минимум U с помощью компьютерной программы поиска стационарных точек на многомерных потенциальных поверхностях. Таким образом, практические действия исследователя чаще всего сводятся только лишь к заданию стартовой геометрии и вызову программы оптимизации геометрических параметров из условия минимума энергии. На выдаче просматривается полученная структура и. если необходимо, анализируются энергия и ее составляющие.
Трудно переоценить роль молекулярной механики в современной химической практике. Поскольку все вычислительные проблемы относятся лишь к хорошо разработанным процедурам минимизации, даже на маломощных персональных компьютерах можно анализировать строение больших многоатомных молекул за разумное время. Возможность увидеть структуру молекулы на экране компьютера, рассмотреть ее с разных сторон, проверить возникающие предположения о стерических затруднениях и т.д. оказывает неоценимую помощь в работе. Молекулярная механика играет роль молекулярного конструктора: для первичной оценки строения интересующей нас молекулы зачастую проще собрать молекулу на компьютере, чем тратить время на поиск необходимой информации в справочной литературе. При расчетах молекулярной структуры на более высоком уровне методами квантовой химии полезно использовать координаты ядер молекулы, найденные с помощью молекулярной механики, в качестве начального приближения. Для многих задач, например для конформационного анализа, уровень моделирования методами молекулярной механики оказывается вполне достаточным для качественных и даже количественных заключений.
В каждом конкретном случае необходимо интересоваться, для каких классов соединений параметризована та версия программы, которую предполагается применять при моделировании свойств нового соединения. Особенно осторожно следует относиться к оценкам энергий, хотя и для геометрических конфигураций возможны грубые ошибки.[5,6,7]
При моделировании методами молекулярной динамики или Монте-Карло интересующее нас свойство системы большого числа молекул вычисляется через статистические средние по положениям и движениям молекул. Как и в методах молекулярной механики, здесь также необходимо перечислить все частицы системы и задать потенциалы межчастичных взаимодействий. Однако в отличие от молекулярной механики в данных подходах области задания межчастичных потенциалов взаимодействия должны быть достаточно протяженными, и они не должны ограничиваться малыми смешениями от положений равновесия. Это накладывает существенно более высокие требования на способы расчета потенциалов.
Практически всегда уравнения, связывающие молекулярные параметры и свойства вещества, то есть макроскопические свойства, решаются численно, а эффективность решения существенно зависит от мощности используемых компьютеров. На рисунке 2 показаны схемы двух методик: Монте-Карло и молекулярной динамики, применяемых в компьютерных экспериментах. В обоих случаях задаются число молекул N, объем V, доступный для движения молекул, накладываются те или иные граничные условия, предписывается потенциал межмолекулярного взаимодействия U. В методе Монте-Карло обычно независимыми переменными, сохраняющими постоянные значения при моделировании, выбираются N, V и температура Т. Молекулы двигаются случайным образом в соответствии с предписаниями генератора случайных чисел, и каждое новое расположение либо принимается, либо отбрасывается с вероятностью, определенной по закону
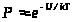 ,
,
где k — константа Больцмана.
Рисунок 2 – Схема расчетов методами Монте-Карло и молекулярной динамики
При моделировании в рамках молекулярной динамики положения r(t) и скорости v(t) каждой частицы в момент времени t определяются как решения системы уравнений классической механики (уравнений Ньютона) либо уравнений, в которых к силам F задаваемым потенциалом U, добавляются так называемые случайные силы. Макроскопические свойства рассчитываются при усреднении по положениям и скоростям молекул.
Как уже упоминалось, число частиц при моделировании методами Монте-Карло и молекулярной динамики с помошью современных суперкомпьютеров может достигать колоссальных величин. Даже без суперкомпьютеров достаточно типичны численные эксперименты для значений N порядка десятков и сотен тысяч. Примеры успешного применения методов Монте-Карло и молекулярной динамики для моделирования равновесных составов смесей при постоянном давлении, фазовых равновесий, адсорбции на поверхности твердых тел, свойств жидкостей в микропорах и т.д. достаточно многочисленны. Этими же методами решаются задачи поиска устойчивых конформаций (поворотных изомеров) полимерных молекул, чрезвычайно важные для биохимических приложений [5,6].
Рассмотрим достаточно последовательную квантовую модель на примере бимолекулярной реакции типа
Х(i) + Y(j) → Х'(i') +Y'(j') + …
Здесь предполагается столкновение двух молекул X и Y, находящихся в состояниях i и j соответственно, которое приводит к продуктам реакции, то есть к молекулам X', Y',... в квантовых состояниях i', j', ... Квантовая теория столкновений в принципе позволяет вычислить вероятности переходов между состояниями, отвечающими реагентам и продуктам, затем найти парциальные, то есть относящиеся к данным наборам квантовых чисел (здесь i, j, i', j',...), константы скорости. При усреднении по квантовым состояниям реагентов и продуктов можно оценить макроскопическую константу скорости соответствующей газофазной химической реакции как функцию температуры.
Полное осуществление этой программы в конкретных приложениях крайне затруднительно, даже если из предшествующих квантово-химических расчетов известна поверхность потенциальной энергии. Самой сложной стадией является численное решение уравнений квантовой теории столкновений с учетом перераспределения частиц, то есть как раз наиболее важная для химии стадия. Следует, однако, подчеркнуть исключительную важность научных исследований в этом направлении, поскольку они формируют каркас обшей теории, с которой сравниваются более простые модели. Кроме того, современные экспериментальные методы исследования динамики молекул позволяют измерить парциальные константы скорости и непосредственно сопоставить экспериментальные и теоретические результаты.
Более простые, а потому и более практичные способы вычисления констант скорости химических реакций получают обычно при определенных упрощениях полной квантовой модели. Так, начиная с 50-х годов проводятся компьютерные расчеты скоростей реакций методом классических траекторий. В этом методе, как и ранее, предполагается разделение электронной и ядерной подсистем, но в данном приложении необходимо знание поверхностей потенциальной энергии для достаточно широких интервалов межъядерных расстояний. Для расчета движений ядер, совместимых с данной потенциальной поверхностью, решают уравнения классической механики, а оценки констант скорости получают при сопоставлении числа траекторий, приводящих к реакции, с исходным числом траекторий при статистическом задании начальных условий.
На рисунке 3 изображены последовательные стадии вычислений методом классических траекторий. Реализация подобной схемы на современных компьютерах позволяет вычислять константы скорости реакций с существенно большим числом атомов, нежели при полностью квантовом описании [5,6].

Рисунок 3 – Схема расчетов констант скоростей химических реакций методом классических теорий
1.3 Определение биологической активности по модели
Для методов определения биологической активности вводится понятие о дескрипторах и QSAR. Молекулярный дескриптор – это числовые значения, характеризующие свойства молекул. Например, они могут представлять физико-химические свойства. Многие различные молекулярные дескрипторы описаны и применяются для различных целей. Они различаются по сложности, закодированной в них информации и сложности расчетов. Увеличение потребности в вычислительной технике увеличивается со сложностью расчетов.
Например, молекулярная масса имеет малое значение среди химических свойств, но зато быстро вычисляется. Дескрипторы основанные на квантово-химических расчетах имеют более важное значение для получения информации о химических свойств, но очень длительны по времени. Дескриптор может быть рассчитан из двухмерной и трехмерной модели химической структуры. Полученные дескрипторы обрабатываются и объединяются. Особое внимание заслуживают дескрипторы, которые описывают свойства молекул, а не замещают их. Такой вид дескрипторов является важной частью в разработке метода QSAR.
Большое распространение имеют математические и статистические модели. К таким моделям относятся модели методов QSAR(определяет количественные соотношения между структурой и активностью) и QSPR(определяет количественные соотношения между структурой и свойствами). Модель выполненная по методу QSAR должна быть разработана, как «эксперимент» и соответствовать точности реального эксперимента, для адекватного прогноза и для получения максимальной пользы от модели. В данном случае, чем больше первоначальных данных, тем точнее модель. Первым этапом является определение размера набора данных. Вторым этапом является корреляция дескрипторов. После определения набора данных и некоррелированных дескрипторов решается что именно должно быть включено в уравнения QSAR. Самый простой способ это использование автоматизированной процедуры. Полученные дескрипторы просчитываются через ряд уравнений и по полученным значениям определяют активность.
Последнее время одна из наиболее важных разработок с применением метода QSAR в области биологической активности связано с введением в CoMFA (Comparative Molecular Field Analysis). Цель CoMFA заключается в связывании биологической активности с трехмерной формой молекулы, электростатическими характеристиками и водородными связями. Структура данных используемая в анализе CoMFA вытекает из ряда комформаций, по одной на каждую молекулу. По этим конформациям и просчитывают биологическую активность молекулы.
Просеивание с высокой пропускной способностью (HTS-метод). Сегодня HTS-метод (High Throughput Screening) повсеместно используется в фармацевтической индустрии для открытия новых лекарственных средств. С помощью высокоскоростной компьютеризованной технологии сотни тысяч веществ проверяются на активность относительно исследуемой молекулы, предназначенной для взаимодействия[8].
2. Применение компьютерного моделирования в современной практике
В США была разработана технология компьютерного моделирования новых химических веществ. Создано специализированное программное обеспечение и разработана обучающая литература по нему. Примером может служить книга Хинклифа «Моделирование молекулярных структур», выпущенная в 2000 году, в котором описаны методы расчетов и моделирования, используемые в HyperChem.
В России компьютерное моделирование находится на начальном этапе развития и его продвигают ученые-энтузиасты. Например, Николай Серафимович Зефиров — химик-органик; академик РАН. Зефиров внёс вклад в математическую химию, в решение проблемы описания органических структур и реакций; проблемы «структура-активность» (QSAR), проблемы поиска структуры, отвечающей заданному целевому свойству (QSPR); в компьютерное моделирование и компьютерный синтез. Занимался поиском новых реакций и реагентов, созданием методов синтеза целевых структур; соединений-лигандов глутаматных, мелатониновых и других рецепторов как потенциальных лекарств[9].
Достижения Зефирова в последние годы кратко можно сформулировать следующим образом:
1) развиты методы математической химии, на основе которых было осуществлено молекулярное моделирование строения и функционирования ряда важнейших рецепторов человека и компьютерный дизайн их потенциальных лигандов; получен патент на новый лекарственный препарат для лечения болезни Альцгеймера, принципиально отличающийся по принципу действия от всех описанных ранее препаратов;
2) проведенные молекулярно-динамические расчеты лиганд-рецепторных комплексов и свободных форм рецепторов позволили предсказать и объяснить агонистподобное расположение антагонистов в лиганд-связывающих центрах рецепторов, важное функциональное значение димеризации аминоконцевых доменов, возможность моделирования процессов закрытия и открытия аминоконцевого домена, а также предположить альтернативное объяснение функциональной роли агонистов, заключающееся в изменении конформаций боковых цепей аминокислотных остатков. С помощью метода молекулярного моделирования впервые построены полные пространственные модели серии рецепторов человека, что позволяет решать задачи конструирования и поиска лекарств по типу мишени (рецептора, на который данное лекарство действует);
3) созданы методы молекулярного докинга для исследования лиганд-белковых взаимодействий в белковых структурах, что позволяет осуществить предсказание связывания химических соединений с рецепторами на основании виртуального эксперимента. Выявлены соединения-лидеры с уникальным спектром нейропротекторных и когнитивно-стимулирующих свойств.
Зефиров заведуют лабораторией математической химии и компьютерного синтеза Института органической химии им. Зелинского РАН[10].
В КГМУ где ведется подготовка специалистов в области синтеза биологически активных веществ выпускающая кафедра биологической и химической технологии стала в ряды энтузиастов молекулярного моделирования. Далее приводятся примеры работ выполненные на кафедре биологической и химической технологии.
3. Примеры применения молекулярного моделирования
3.1 Определение механизма взаимодействия медиатора и рецептора с использованием молекулярного моделирования на примере ГАМК
Пытаясь объяснить роль ГАМК в нервной системе встречается одна из центральных проблем - выяснением механизма передачи импульсов в нервной системе. В ткани головного мозга содержатся химические вещества (медиаторы, передатчики), которые участвуют в создании контактов между нервными клетками осуществляют перенос импульса как в периферической, так и в центральной нервной системе. Решение этой центральной проблемы в настоящее время тесно связано с детальным изучением пространственного строения взаимодействующих медиаторов и рецепторов.
Взаимодействие ГАМК с нервной клеткой осуществляется когда нервный импульс достигает нервного окончания, он вызывает освобождение медиатора в синаптическую щель. ГАМК диффундирует через синаптическую щель (шириной 20—50 нм) и взаимодействует со специальными рецепторами в постсинаптической мембране. В результате такого взаимодействия изменяется мембранная проводимость для ионов, находящихся по обе стороны мембраны. Изменение проводимости связано со структурными изменениями мембраны и открытием в ней узких гидрофильных каналов для прохождения катионов или анионов и зависит от структурных особенностей ГАМК.
ГАМК имеет неплоскую структуру. ГАМК является гибкой цвиттер-ионной молекулой, которая может существовать в разных конформациях. Методами рентгеноструктурного анализа, изучением действия синтетических аналогов с жестко фиксированным строением и ряда природных агонистов и антагонистов ГАМК было надежно установлено, что наиболее предпочтительной для получения тормозящего эффекта является вытянутая конформация ГАМК (расстояние между заряженными атомами N+ и О- составляет в этом случае 5,4 ± 0,4 А; для свернутой конформации оно находится в пределах 3,9-4,2 А).
В кристаллах ГАМК находится полностью в вытянутой конформации. Однако в растворах ее молекулы, вероятно, существуют в различных конформациях. Прямое экспериментальное определение структуры ГАМК в момент ее взаимодействия с рецептором пока невозможно.
Для установления пространственного строения активной молекулы ГАМК и участка ее рецептора была изучена связь структуры с физиологической активностью некоторых аналогов ГАМК, имеющих в отличие от гибкой молекулы ГАМК жесткую структуру. Так, мусцимол, 4-аминотетрловая и 4-аминокротоновая кислоты имеют определенную конформацию благодаря наличию циклической структуры двойных и тройных связей. Различное действие мусцимола и других аналогов ГАМК, на спинные нейроны, а также структурные характеристики ряда ингибиторов захвата ГАМК показывают, что в процесс поглощения ГАМК нервными структурами и взаимодействия ее с постсинаптическими рецепторами она может иметь как вытянутую, так и свернутую конформацию.
Методами молекулярного моделирования были получены модели молекулы ГАМК и ГАМК-рецептора. Модель молекулы ГАМК была получена в программе HyperChem v6.0 методом оптимизации PM3. Пример модели молекулы ГАМК приведен на рисунке 4.
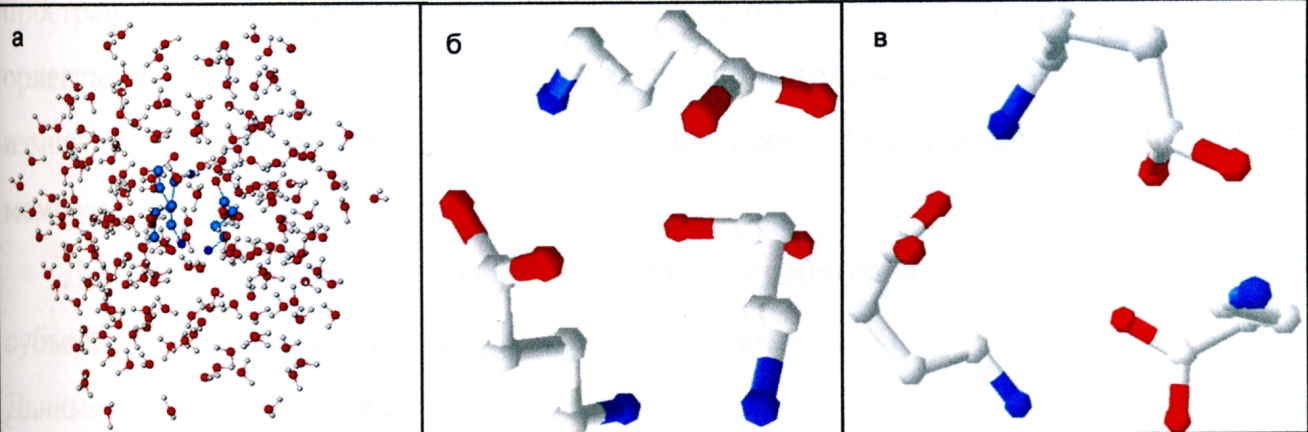
Рисунок 4 – Модели молекулы ГАМК. а) оптимизация молекул ГАМК в воде; б) вытянутая конформация; в) свернутая конформация.
Была выдвинута гипотеза, что взаимодействие молекулы ГАМК и рецептора обусловлено конформацией молекулы ГАМК. Положение ионов N+ и О- и расстояние между ними обуславливает образование комплекса между рецептором и медиатором. Образованный комплекс изменяет проводимость мембраны за счет образования пор.
Для проверки гипотезы была построена модель рецептора по аминокислотной последовательности, полученной из базы данных RCSB.PDB (Protein Data Bank), в специализированной программе DeepView – TheSwissPdbViewer v3.7. Построенная модель приведена на рисунке 5.

Рисунок 5 - Модель рецептора ГАМК.
Как видно из модели ГАМК-рецептора в центре есть канал для связывания с молекулами ГАМК. Размеры канала составляют 6,65 Ао, а размер комплекса из трех молекул вытянутой конформации ГАМК составляет 6,012 Ао, что позволяет предположить что взаимодействие происходит именно поэтому месту [11].
3.2 Определение биологической активности при помощи программы PASS
Объектом является химическое соединение – дикаин.
Структурная формула дикаина:
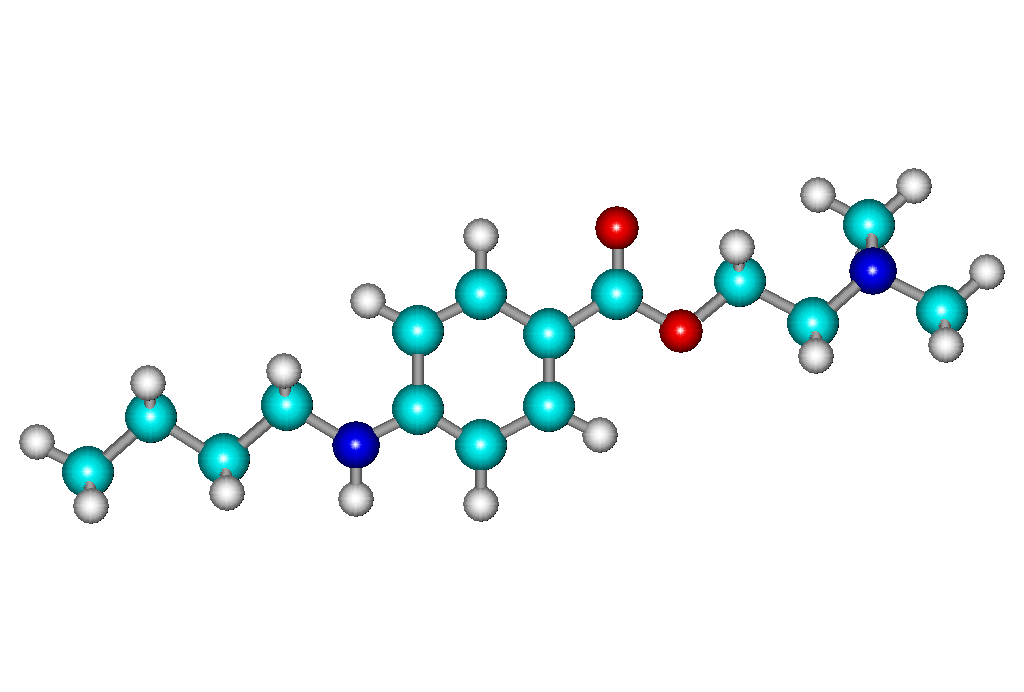
Температура
плавления 147-150 .
Соединение представляет белый
кристаллический порошок без запаха.
Легко растворим в воде и спирте. Дикаин
сильное местноанестезирующее средство,
обладающее высокой токсичностью.
Применяют в глазной и оториноларингологической
практике при некоторых оперативных
вмешательствах, а также для анестезии.
.
Соединение представляет белый
кристаллический порошок без запаха.
Легко растворим в воде и спирте. Дикаин
сильное местноанестезирующее средство,
обладающее высокой токсичностью.
Применяют в глазной и оториноларингологической
практике при некоторых оперативных
вмешательствах, а также для анестезии.
Были предложены варианты новых структур для компьютерного дизайна молекулы дикаина с целью снижения его токсичности с сохранением или даже усилением анестезирующих свойств.
Введение в бензольное кольцо «облагораживающей» карбоксильной группы и замена диметиламиногруппы на более фармакоактивную диэтиланиногруппу позволит снизить токсичность соединения, облегчить гидролиз сложноэфирной связи с высвобождением антигистаминного фрагмента - диэтиламиноэтанола.
Алифатический радикал н-бутил в структуре дикаина усиливает фармакологический эффект. При замене его на адреналиновый фрагмент ожидается получить более яркое анестезирующее действие.
К настоящему времени известно, что биологические системы не делают различия между плоскими кольцами, поэтому при замене ароматической основы н-аминобензойной кислоты на никотиновую (или изоникотиновую) кислоту изменяется полярность молекулы, облегчается задача введения различных заместителей в ароматическое кольцо. К тому же, аминопроизводные никотиновой кислоты (кордиамин) являются стимуляторами центральной нервной системы.
Один из наиболее эффективных анестетиков, промедол, содержит в структуре вместо ароматического пиридинового кольца пиперидиновое, что является предпосылкой для модификации молекулы дикаина.
Условно новым структурам дали названия:
1) Структура 1 – карбоксиструктура;
2) Структура 2 – адреноструктура;
3) Структура 3 – никотиноструктура;
4) Структура 4 – пиперидиноструктура.
Примеры структур приведены на рисунке 6.
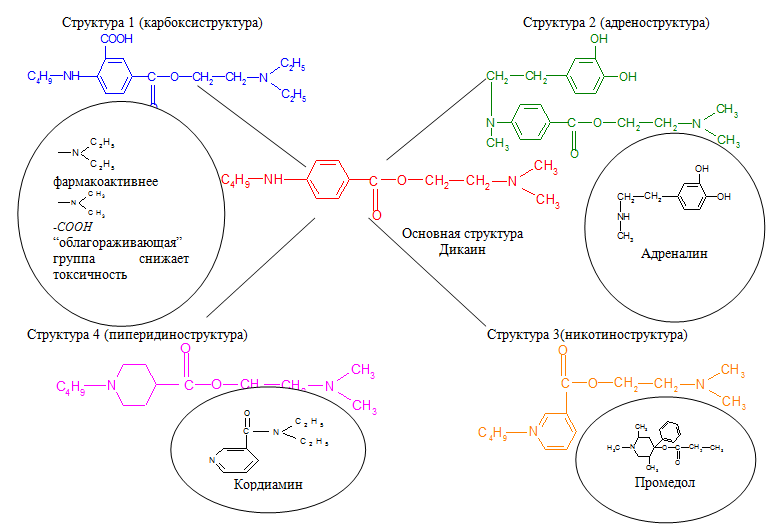
Рисунок 6 – Предложенные структуры.
При помощи программы HyperChem была доказана возможность существования данных соединений путем оптимизации молекул и построения графиков потенциальной энергии. По графикам были обнаружены максимумы и минимумы, что подтверждает существования молекулы по квантовым расчетам.
После доказательства существования молекулы была проведена проверка биологической активности на программе PASS. Сводный анализ биологической активности дикаина и новых структур приведены в таблице 1.
Современная версия компьютерной системы предсказания спектра биологической активности PASS C&T (Prediction of Activity Spectra for Substances: Complex & Training) реализована в 1998 году. Она включает в себя обучающую выборку, содержащую более 45000 биологически активных веществ с известной биологической активностью, и охватывает более 400 фармакологических эффектов, механизмов действия, а также мутагенность, канцерогенность, тератогенность и эмбриотоксичность[12].
Таблица 1 Таблица биологической активности ряда веществ
|
Характеристика фармакологической активности |
Основная структура |
Модифицированные структуры |
|||
|
Дикаин
|
Структура 1 Карбоксиструктура
|
Структура 2 Адреноструктура
|
Структура 3 Никотиноструктура
|
Структура 4Пиперидноструктура
|
|
|
1. Спазмолтик |
0,603 0,023 |
0,591 0,025 |
0,620 0,021 |
0,683 0,017 |
0,680 0,015 |
|
2. Сосудорасширяющее средство |
0,511 0,048 |
0,367 0,095 |
0,472 0,059 |
0,548 0,031 |
0,537 0,042 |
|
3. Антагонист кальциевых каналов |
0,405 0,015 |
0,264 0,051 |
0,362 0,020 |
0,326 0,026 |
0,411 0,014 |
|
4. Антигипер-тензивный |
0,350 0,107 |
0,301 0,142 |
0,237 0,211 |
0,364 0,098 |
0,402 0,078 |
|
5. Агонист β-адренорецепторов |
0,114 0,098 |
0,113 0,101 |
0,139 0,043 |
||
|
6. Токсичный |
0,323 0,166 |
0,331 0,160 |
0,291 0,188 |
||
|
7. Тератоген |
0,219 0,214 |
0,233 0,195 |
0,243 0,182 |
||
|
8. Антагонист β-адрено-рецепторов |
0,092 0,091 |
0,092 0,09 |
|||
|
9. Диуретик |
0,211 0,144 |
||||
|
10. агонист
|
0,144 0,041 |
||||
|
11. Агонист α-адренорецепторов |
0,133 0,119 |
0,253 0,075 |
|||
|
12.
Антагонист
|
0,233 0,051 |
 -адренорецепторов
-адренорецепторов -адренорецепторов
-адренорецепторов3.3 Поиск физиологически активных аналогов ССК-4
Было известно, что многие пептиды, в частности, аналоги гастроинтерального гормона холецистокинина(ССК), состоящего из 33 аминокислот (ССК-33), при парентеральном введении обладают способностью существенно влиять на психоэмоциональный статус. ССК пептиды функционируют в центральной нервной системе(ЦНС) как нейромодуляторы и участвуют в регуляции поведенческих реакций, болевых ощущений, процессов памяти и обучения.
В связи с этим следует ожидать, что модуляция ССК системы является основой для лечения и предупреждение психотических заболеваний.
Наиболее перспективным направлением при поиске средств купирования психических расстройств является конструирование аналогов эндогенных пептидов, который характеризуются минимальной токсичностью и отсутствием побочных эффектов. В этом аспекте актуальны разработка и синтез соединений на основе нейропептидов – антагонистов минимального физиологически активного С-концевого фрагмента холецистокинина – пептида ССК-4. ССК-4 - селективный агонист к холецистокининовым рецепторам второго типа, которые вовлечены в формирование психоэмоционального состояния.
Пространственная структура рецептора была получена из базы данных Protein Data Bank (www.spdb.org). Оптимизацию молекул проводили с применением метода молекулярной механики BIO+ и полуэмпирическим квантово-химическим методом PM3, которые реализованы в программном комплексе HyperChem. В итоге по результатам молекулярного моделирования были получена наиболее вероятная биологически активная конформация молекулы ССК-4, которая представлена на рисунке 7.
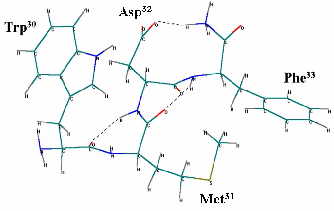
Рисунок 7 – Биологически активная конформация ССК-4
Исходя из структуры биологической конформации были предложены аналоги. В ходе данной работы были получены следующие результаты:
Методами молекулярного моделирования сконструирована вероятная биологически активная конформация пептида ССК-4, а также разработана пространственная модель взаимодействия лиганда ССК-4 с активным центром холецистокининового рецептора второго типа.
С использованием квантохимических расчетов разработанной модели взаимодействия пептида ССК-4 и холецистокининового рецептора второго типа предложен вероятный механизм лиганд-рецепторного взаимодействия, заключающийся в одновременном переносе протона амидной группы N-концевой группы фенилаланина и перераспределением электронной плотности с молекулы лиганда ССК-4 на аминокислоту активного центра рецептора.
На основании рассчитанных дескрипторов предложены новые высокоактивные соединения – аналоги ССК-4.
Синтезированы предложенные аналоги.
Проведены экспериментальные исследования по оценке биологической активности синтезированных аналогов. Установлено, что аналоги превосходят стандарт в 3,6-4,1 раза по антагонистической активности [13].
4. Анализ примеров
Анализ приведенных примеров позволяет предложить следующую схему использования компьютерного моделирования в создании новых лекарственных препаратов:
Анализ литературы по фармацевтической химии и синтезу лекарственных препаратов. Поиск моделей структур соединений и рецепторов в базах данных, полученных методами ЯМР и рентгеноскопии. Примерами баз данных служат SPDB, CSD.
В случае отсутствия компьютерных моделей препарата строят модель и оптимизируют модель квантово-химическими расчетами в программном комплексе HyperChem или аналогичном;
Анализ биологической активности путем совмещения модели рецептора клетки-мишени или путем использования метода QSAR в программе PASS;
По сделанным выводам о биологической активности предлагают модификации, улучшающие свойства молекулы. При этом должна использоваться литература по фармацевтической химии и синтезу лекарственного препарата;
Построение модели в HyperChem, анализ биологической активности методами виртуального докинга или QSAR с использованием HyperChem и PASS, соответственно;
Синтез новой структуры и лабораторные испытания.
Заключение
В ходе данной работы было проанализированы работы о применимости методов моделирования в практике, а именно при создании новых препаратов и изучении механизма действия химического соединения и рецепторов клетки мишени.
По результатам анализа можно с уверенностью сказать, что молекулярное моделирование является эффективным методом разработки новых лекарственных препаратов, позволяющим сэкономить время при отборе новых структур и деньги, затрачиваемые на синтез новых структур для отбора. Но применение данной методики связано с определенными трудностями, так как требует высококвалифицированных кадров. Также это требует высокие вычислительные мощности оборудования и соответствующее программное обеспечение, которое требует больших финансовых затрат.
Список используемых источников
Ассоциация Российских Фармацевтических производителей. Режим доступа: http://www.arfp.ru, свободный;
ЦВТ «Химрар». Режим доступа: http://www.chemrar.ru, свободный;
A.R. Leach, V.J. Gillet: An Introduction to Chemoinformatics. Springer, 2003, ISBN 1-4020-1347-7;
A.R. Leach, V.J. Gillet: An Introduction to Chemoinformatics. Springer, 2003, ISBN 1-4020-1347-7;
Степанов Н.Ф. Квантовая механика и квантовая химия. – М.: Мир, 2001. – 519 с., ил;
Немухин А.В. Компьютерное моделирование в химии // Соросовский образовательный журнал, 1998. — №6. — с.48-52;
9. Буркерт У., Аллинжер Н. Молекулярная механика. М.: Мир, 1986. - 364 с;
Курс лекций «Введение в химическую информатику», проведенный в 2008 г. в Киевском политехническом институте. Режим доступа: http://summerschool.ssa.org.ua/index.php?option=com_content&task=view&id=91&lang=ru , свободный;
Зефиров, Николай Серафимович – Википедия. Режим доступа: http://ru.wikipedia.org/wiki/Зефиров, свободный;
Биографическая интернет-энциклопедия. Режим доступа: http://www.biographica.ru/, свободный;
Дипломный проект А.В.Конорева «Производство ГАМК мощностью 7 тонн в год в системе фармпредприятий. Исследование механизма рецепции ГАМК методом компьютерного моделирования»;
Дипломный проект Саенко «Разработка аналогов дикаина методами компьютерного моделирования»;
Автореферат диссертации С.В.Шульгина на тему «Конструирование синтетических аналогов С-концевого тетрапептида холецистокинина и оценка их физиологической активности»