Определение примесей в технических целлюлозах
РЕФЕРАТ
На тему
Определение примесей в технических целлюлозах
Москва, 2009
Введение
Присутствие в технической целлюлозе различных примесей, таких, как минеральные вещества, смолы и жиры, остаточный лигнин и пентозаны, характеризует степень ее чистоты и определяет возможность дальнейшей переработки целлюлозы в различные ее производные. Все нецеллюлозные примеси, по существу, являются нежелательными и количество их в целлюлозе для химической переработки должно быть минимальным. Отрицательное их влияние на технологический процесс переработки и свойства готовых искусственных волокон и пленок изучено и освещено в литературе достаточно подробно.
Зольность целлюлозы существенно влияет на процессы получения вискозных и ацетатных волокон и пленок. Даже незначительное повышение общего содержания золы в целлюлозе приводит к нарушению режима производства эфиров целлюлозы и ухудшению свойств готовой продукции. При использовании целлюлозы для химической переработки имеет значение массовая доля золы, но и ее состав.
Ионы металлов переменной валентности оказывают сильное влияние на окислительную деструкцию целлюлозы во время предсозревания щелочной целлюлозы в процессе получения вискозного волокна. Соединения кальция и кремния ухудшают процесс фильтрации вискозы. Кроме того, соли кальция способствуют увеличению засоряемости фильер вследствие образования гипса во время формования волокна, что приводит к обрыву нити.
Реакционная способность целлюлозы при ацетилировании также в значительной степени зависит от содержания в ней различных солей. Даже небольшие количества сульфатов, особенно сульфата магния, присутствующих в целлюлозе, снижают скорость ацетилирования, тогда как силикаты и другие соли марганца ускоряют реакцию. Катионы натрия, железа и меди вызывают помутнение растворов ацетатов целлюлозы. В присутствии зольных элементов происходят заметные изменения и в растворах нитратов целлюлозы. Например, щелочноземельные и поливалентные металлы повышают вязкость концентрированных растворов. Вследствие вышеизложенного в целлюлозах для химической переработки содержание золы согласно требованиям стандартов не должно превышать 0,07...0,12%.
Содержание золы в беленых и небеленых целлюлозах, идущих на производство бумаги и картона, не нормируется, за исключением целлюлозы для выработки электроизоляционных бумаги и картонов. Присутствие в целлюлозах солей, являющихся сильными электролитами, понижает диэлектрические свойства целлюлозы, что имеет отрицательное значение при ее использовании для электроизоляционных видов бумаги. Зольность целлюлозы для электроизоляционных бумаги и картона не должна превышать 0,25 и 0,51%, соответственно.
Повышенное содержание смол и жиров в целлюлозе, применяемой для получения вискозного волокна, нежелательно по нескольким причинам. Прежде всего эти примеси вызывают увеличение содержания воздуха в вискозе, повышают стабильность его пузырьков и тем самым затрудняют процесс их удаления из вискозы. Кроме того, жиры и смолы засоряют отверстия фильер, а при сушке волокна разлагаются и остаются на нем в виде желтых пятен. Однако в литературе имеются и противоположные мнения. Отмечается, что вискоза, полученная из целлюлозы, не содержащей смол и жиров, труднее фильтруется. Поэтому остаточная массовая доля смолы в целлюлозе для вискозообразования должна быть не менее 0,06%, но и не более 0,3%. В процессе ацетилирования содержащиеся в целлюлозе смолы и жиры в значительной мере определяют желтую окраску ацетата целлюлозы.
Лигнин, остающийся в целлюлозе, как правило, понижает ее реакционную способность, ухудшает ее набухание и растворимость полученных эфиров. С увеличением содержания остаточного лигнина замедляется окислительный распад макромолекул целлюлозы в процессе предсозревания щелочной целлюлозы, повышается жесткость искусственных волокон.
Большое влияние как на свойства целлюлозы, так и на качество искусственных волокон и пленок оказывают гемицеллюлозы. Вследствие меньшей длины цепей они легко переходят в раствор при мерсеризации и загрязняют щелочь, а это влечет за собой необходимость ее очистки и, следовательно, усложнение технологического процесса. Маннаны и ксиланы, несмотря на более высокую скорость ацетилирования по сравнению с целлюлозой, образуют ацетаты с низкой растворимостью, что приводит к помутнению растворов ацетатов целлюлозы. Гемицеллюлозы влияют и на вязкость растворов ацетатов целлюлозы.
Таким образом, определение примесей целлюлозы является одним из необходимых условий для характеристики целлюлозы для химической переработки.
1. Определение содержания золы
Количество золы в целлюлозе характеризует содержание в ней минеральных веществ, состав которых обусловлен используемой древесиной, солями жесткости воды, применяемой в технологическом процессе варки и отбелки, механическими загрязнениями из растворов химикатов и используемого оборудования. Метод определения общего содержания золы основан на сжигании и прокаливании навески целлюлозы при определенной температуре до полного удаления углерода. Содержание отдельных элементов в полученной золе целлюлозы определяют с помощью химического и спектрального методов.
Спектральным методом определяют содержание катионов в золе целлюлозы, предназначенной для химической переработки и производства электротехнической бумаги. Сущность метода заключается в сжигании пробы исследуемой золы целлюлозы в дуге переменного тока и фотографировании спектра излучения на фотопластинку. По интенсивности линий спектра определяют содержание каждого элемента золы. Спектральный метод определения элементов золы в сравнении с химическим методом значительно ускоряет проведение анализа, позволяет получать более точные результаты и в настоящее время широко применяется в технологическом контроле ЦБП.
При определении содержания золы в целлюлозе целлюлозу измельчают на кусочки без применения ножей и другой металлической аппаратуры, так как остающиеся на волокне мельчайшие частицы металла могут исказить результаты. Такое условие подготовки образца для анализа особенно необходимо соблюдать при дальнейшем определении составляющих компонентов золы. Сжигание целлюлозы следует проводить осторожно при возможно более низкой температуре, чтобы избежать воспламенения и возможной потери золы.
Методика анализа. Для анализа используют воздушно-сухую измельченную вручную целлюлозу. Масса навески целлюлозы зависит от ее предполагаемой зольности. Навеску берут с таким расчетом, чтобы масса золы составляла 10...20 мг. Чем ниже зольность целлюлозы, тем больше должна быть масса навески:
Навеску целлюлозы определенной массы помещают в предварительно прокаленный и взвешенный тигель и осторожно обугливают в муфельной печи при температуре около 300°С, не допуская воспламенения целлюлозы. Если навеска не помещается сразу в тигель, то ее сжигают частями. Затем золу прокаливают в течение 3 ч при температуре °С до исчезновения темных частиц углерода. Тигель с золой извлекают из муфельной печи с помощью тигельных щипцов, несколько секунд охлаждают на воздухе на несгораемой подставке и помещают в эксикатор. Охлажденный до комнатной температуры тигель с минеральным остатком взвешивают. Прокаливание тигля с золой повторяют по 1 ч до получения постоянной массы.
Массовую долю золы, % к абсолютно сухой целлюлозе, рассчитывают по формуле
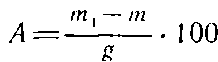
где т — масса пустого тигля, г; ш, — масса тигля с золой после прокаливания, г; g — абсолютно сухая навеска целлюлозы, г.
2. Определение смол и жиров
Массовая доля смол и жиров в целлюлозе, предназначенной для химической переработки, колеблется в пределах от 0,06 до 0,07% для сульфатной предгидролизной и от 0,2 до 0,3% для сульфитной. Состав смол и жиров в целлюлозе зависит как от методов варки и отбелки, так и от исходной древесины. Целлюлоза, полученная из древесины хвойных пород, содержит большее количество смолистых веществ, чем целлюлоза из древесины лиственных пород. Однако в целлюлозах из лиственных пород, особенно в березовой технической целлюлозе, содержатся экстрактивные нейтральные соединения, которые подобно смолистым веществам целлюлозы из хвойных пород вызывают затруднения в технологических процессах получения и переработки производных целлюлозы.
Сущность метода определения смол и жиров в целлюлозе, как и для древесины, заключается в многократном экстрагировании волокнистого полуфабриката органическими растворителями с последующей отгонкой растворителя, сушкой и взвешиванием. В зависимости от применяемого растворителя результаты определения массовой доли смол и жиров в целлюлозе будут различными. До настоящего времени в разных странах для более полного выделения смол и жиров используют смесь двух растворителей или последовательное экстрагирование двумя или тремя растворителями. Экстрагирование целлюлозы осуществляют в аппарате Сокслета.
По ГОСТ 6841—77 для определения содержания смол и жиров в целлюлозе в качестве растворителя используют дихлорметан. Температура кипения дихлорметана равна 40°С. Дихлорметан трудногорючая жидкость, не взрывоопасен в смеси с воздухом, но токсичен. Работу с дихлорметаном следует проводить в вытяжном шкафу.
Методика анализа. Навеску массой около 20. г воздушно-сухой целлюлозы, предварительно нарезанной на кусочки 1,0X 1,0 см, помещают в насадку для экстрагирования вместимостью 150 см. В сифонную трубку насадки предварительно вводят небольшое количество обессмоленной ваты, смоченной дихлорметаном. При этом уровень навески целлюлозы должен быть на 1,0...1,5 см ниже уровня сифонной трубки. В предварительно высушенную и взвешенную до постоянной массы колбу аппарата наливают дихлорметан в количестве, в 1,5 раза превышающем объем насадки Соединяют насадку с холодильником и колбой, ставят аппарат на нагревательную плитку с закрытым электрообогревом. Перед включением электрообогрева пускают в холодильник воду со скоростью, обеспечивающей полную конденсацию паров растворителя. Экстрагирование проводят в течение 3 ч. По окончании экстрагирования отгоняют через экстрактор чистый растворитель до тех пор, пока в колбе не останется 5...7 экстракта. Колбу с экстрактом сушат в сушильном шкафу при температуре °С в течение 3...4 ч с последующим доведением до постоянной массы.
Массовую долю смол и жиров, %, вычисляют по формуле

где т — масса сухой колбы, г; /тг, — масса колбы с экстрактом после сушки, r; g — масса абсолютно сухой навески целлюлозы, г.
Разность между результатами двух параллельных определений не должна превышать 30% среднего значения.
По полученному результату рассчитывают коэффициент экстрагирования
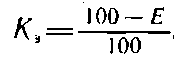
который используют при расчете массовой доли лигнина, неоднородности целлюлозы по молекулярной массе и др.
3. Определение остаточного лигнина
Технические целлюлозы как небеленая, так и отдельные виды беленой содержат некоторое количество остаточного лигнина. Последний представляет собой смесь лигнина и продуктов его реакций, остающихся в технической целлюлозе после делигнификации древесного сырья при варке и после отбеливания небеленой целлюлозы. Остаточный лигнин в целлюлозах, особенно в беленых, является сильно видоизмененным продуктом и количественное определение его представляет собой трудную задачу.
Все методы, используемые для определения остаточного лигнина в целлюлозе, можно подразделить на прямые и косвенные.
Прямые методы основаны на гидролизе целлюлозы концентрированными кислотами. Однако эти методы при использовании для- технических целлюлоз имеют ряд существенных недостатков: длительность и трудоемкость анализа и значительная часть лигнина растворима в кислоте. На точность результатов определения лигнина в целлюлозе оказывают влияние условий гидролиза; природа самой целлюлозы и предварительная ее подготовка к анализу.
Одной из причин, приводящей к ошибкам определения массовой доли остаточного лигнина в целлюлозе, является гумификация углеводов в процессе гидролиза. Продукты гумифиции более легко образуются при гидролизе лиственной целлюлозы по сравнению с хвойной. Поэтому продолжительность гидролиза при определении лигнина в лиственной целлюлозе должна быть несколько уменьшена.
Массовая доля кислоторастворимого лигнина изменяется в зависимости как от применяемого метода, так и от вида исходной целлюлозы. При анализе сульфитных целлюлоз образуется больше кислоторастворимого лигнина, чем при определении лигнина в сульфатных целлюлозах. Это объясняется различным химическим составом лигнина, образующегося в разных процессах варки.
Большое значение при определении остаточного лигнина имеет предварительное измельчение целлюлозы. Отсутствие в целлюлозной массе комочков и узелков позволяет быстрее проводить гидролиз и получать воспроизводимые результаты. Необходимым условием для определения лигнина, как и в случае анализа древесины, является предварительное удаление смол и жиров путем экстрагирования органическими растворителями. Следует подчеркнуть, что при этом происходит ускорение процесса гидролиза целлюлозы в результате увеличения ее доступности к действию минеральных кислот.
Несмотря на отмеченные недостатки, прямые методы определения остаточного лигнина в целлюлозах, особенно в небеленых, до сих пор широко применяются в научно-исследовательских лабораториях. Предложен ряд модифицированных методов, которые способствуют более быстрому и полному гидролизу и меньшей гумификации образовавшихся Сахаров. Так, для определения остаточного лигнина в небеленой целлюлозе чаще всего применяют метод, основанный на гидролизе целлюлозы смесью концентрированных соляной и серной кислот. Этот метод позволяет более быстро проводить анализ и получать воспроизводимые результаты. Однако для получения точных результатов необходимо учитывать и кислоторастворимый лигнин, который определяют УФ-спектрофотометрическим методом так же, как и при анализе древесины.
Для контроля процессов варки и отбелки целлюлозы на практике наиболее часто используют косвенные методы, в которых определяется не массовая доля остаточного лигнина, а степень делигнификации целлюлозы, косвенно указывающая на содержание лигнина. Степень делигнификации — это показатель качества целлюлозы, характеризуемый долей лигнина, удаленного при делигнификации. Иногда рассчитывают относительную степень делигнификации целлюлозы как отношение массы лигнина, удаленного при делигнификации, к массе исходного лигнина в сырье и выражают это отношение в процентах. В основе косвенных методов лежит использование свойства легкой окисляемости лигнина при действии различных специфических окислителей — хлора, брома, перманганата калия, которые разрушают лигнин в условиях анализа, но не воздействуют на целлюлозу. Результаты определения выражают в виде хлорных, бромных и перманганатных чисел, характеризующих так называемую жесткость целлюлозы. Жесткие целлюлозы содержат много остаточного лигнина, мягкие мало. Косвенные методы используют только для волокнистых полуфабрикатов с выходом не более 70%. Они малоточны для беленых целлюлоз с очень низким содержанием остаточного лигнина и не применимы к древесным и термомеханическим массам.
В настоящее время наибольшее распространение во всех странах получил метод определения жесткости целлюлозы по ресманганафному числу.
Методика анализа для небеленой целлюлозы. Анализируемую целлюлозу предварительно измельчают на кусочки 1 X 1,5 мм и экстрагируют дихлорметаном в аппарате Сокслета в соответствии с методикой определения смол и жиров. Из обессмоленной воздушно-сухой целлюлозы берут навеску массой около 1 г и помещают в колбу с притертой пробкой вместимостью 500 см3.
Влажность обессмоленной целлюлозы определяют предельной пробе. В колбу с навеской заливают соляной кислоты и ставят па 30 мин в термостат или водяную баню °С, периодически встряхивая в течение 1 мин через каждые 5..6 мин во избежание образования комков. Затем содержимое охлаждают до комнатной температуры и приливают 90 см3 72%-ной серной кислоты. Смесь выдерживают в течение 1,5 ч при температуре 0С, встряхивая через каждые 10... 15 мин. По истечении этого времени содержимое колбы разбавляют 150 см1 дистиллированной воды. Полученный раствор доводят до кипения, кипятят 1,5...2 мин, а затем содержимое колбы охлаждают и фильтруют. Фильтрование следует проводить на следующий день для укрупнения частиц и облегчения фильтрования. Раствор с осадком лигнина фильтруют через сложенные вместе два уравновешенных на аналитических весах бумажных фильтра. Осадок лигнина и фильтры тщательно промывают горячей дистиллированной водой до полного удаления кислоты. Наличие кислоты в промывных водах и краях фильтров проверяют индикатором метиловым оранжевым.
Фильтры с лигнином сушат в сушильном шкафу при температуре 0C до постоянной массы и взвешивают верхний фильтр с лигнином, помещая нижний фильтр на чашку аналитических весов с разновесами.
Массовую долю кислотонерастворимого лигнина, % к абсолютно сухой необессмоленной целлюлозе, рассчитывают по формуле
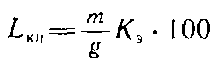
где т — масса лигнина, г; g — масса навески абсолютно сухой обессмоленной целлюлозы, г; K-,—коэффициент экстрагирования.
Разность между результатами двух параллельных определений не должна превышать 0,5%.
Методика анализа для беленой целлюлозы. Из предварительно измельченной и обессмоленной воздушно-сухой целлюлозы берут навеску массой около 1 г, помещают в сухой стакан вместимостью 500 см3. Целлюлозу смачивают 10 см3 дистиллированной воды и через 10 мин охлаждают в бане с проточной водопроводной холодной водой. К содержимому стакана приливают мерным цилиндром 25 см3 86%-ной H2SO4. Во избежание деструкции целлюлозы кислоту добавляют небольшими порциями, тщательно перемешивая стеклянной палочкой и не допуская разогрева. Затем стакан выдерживают в термостате в течение 4 ч при температуре 18...22°С, периодически перемешивая содержимое стакана. По окончании растворения целлюлозы в стакан вливают при помешивании 250 см3 дистиллированной воды, смесь нагревают на электроплитке 'с асбестовой сеткой до кипения и кипятят в течение 5 мин. После этого стакан помещают сначала в кипящую водяную баню на 1 ч, а затем в баню с холодной водой на 15 мин. Раствор с осадком фильтруют через предварительно высушенный до постоянной массы стеклянный пористый фильтр, применяя нафталиновую подушку. Осадок промывают дистиллированной водой до исчезновения следов серной кислоты. Фильтр с осадком сушат до постоянной массы и взвешивают. Массовую долю кислотонерастворимого лигнина, % к абсолютно сухой необессмоленной целлюлозе, рассчитывают по формуле
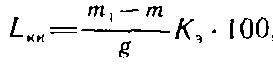
где т — масса стеклянного пористого фильтра, г; m1 — масса стеклянного пористого фильтра с лигнином после сушки, г; g — масса навески воздушно-сухой обессмоленной целлюлозы, г; K>3> — коэффициент экстрагирования.
Разность между результатами двух параллельных определений не должна превышать 30% среднего, значения.
Методика определения кислоторастворимого лигнина. Для определения кислоторастворимого лигнина используют смеси лигнина с кислотой, полученные по вышеуказанным методикам определения кислотонерастворимого лигнина в беленой и небеленой целлюлозах. Смесь переносят в стакан вместимостью 1 дм3, вымывая лигнин из колбы горячей дистиллированной водой в количестве 560 см3. Кипятят лигнин с разбавленной кислотой в открытом стакане на плитке в течение 5 ч. Дальнейший ход анализа и расчет см. 2.9.4. С целью получения более точных результатов при построении градуировочного графика рекомендуется использовать лигнин, осажденный из отработанных варочных щелоков, например щелочной лигнин или лигносульфонаты в случае анализа целлюлоз, полученных соответственно щелочными и сульфитными методами варки, с подбором подходящего растворителя.
Общее содержание лигнина будет равно сумме кислотонерастворимого и кислоторастворимого лигнинов
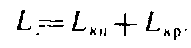
5. Определение жесткости целлюлозы по перманганатному числу
Жесткость целлюлозы по перманганатному числу — это показатель качества, зависящий от содержания остаточного лигнина и определяемый по расходу раствора перманганата калия концентрацией 0,1 моль/дм3 на окисление лигнина в 1 г абсолютно сухой целлюлозы в условиях, установленных стандартом и предусматривающих 50%-ное поглощение перманганата калия. Расход раствора перманганата калия определяют иодометрическим методом
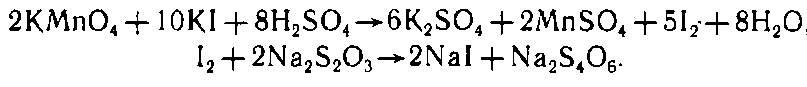
Для образцов целлюлозы с разным содержанием лигнина, чтобы поддерживать %-ное поглощение перманганата калия в конце реакции, подбирают массу навески целлюлозы в пределах от 0,1 до 10 г.
В случае, когда содержание лигнина неизвестно, проводят предварительное определение расхода раствора перманганата калия в соответствии с изложенной ниже методикой анализа. Если он выходит за пределы 30...70%, но находится в интервале 10...80%, массу навески можно рассчитать по формуле
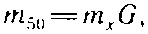
где m>50> — масса абсолютно сухой целлюлозы для 50%-ного поглощения перманганата калия, г; т>х> — масса "абсолютно сухой целлюлозы при х%-ном поглощении перманганата калия, г; G — фактор пересчета.
Фактор пересчета G
|
Поглощение перманганата калия,% |
G при увеличении поглощения KMriO>4 >на 1, 2, 3....% |
|||||||||
|
0 |
I |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
|
10 20 30 40 50 60 70 |
5,69 2,69 1,74 1,27 1,00 0,82 0,70 |
5,13 2,55 1,68 1.24 0,98 0,81 0,69 |
4,67 2,43 1,62 1,21 0,96 0,79 0,68 |
4,29 2,31 1,57 1,18 0,94 0,78 0,67 |
3,96 2,21 1,52 1,15 0,92 0,77 0,66 |
3,67 2,11 1.47 1,12 0,90 0,75 0,65 |
3,42 2,03 1,43 1,09 0,89 0,74 0,64 |
3,21 1,95 1,39 1.07 0,87 0,73 0,63 |
3,01 1,87 1,35 1,05 0,85 0,72 0,62 |
2,84 1,80- 1,31 1,02 0,84 0,71 0,61 |
Ниже приводятся две методики определения жесткости целлюлозы по перманганатному числу — методика ГОСТ 10070— 74 и Международного комитета анализов целлюлозы.
Методика анализа. Массу навески выбирают с учетом вышеприведенных указаний.
А. Подготовленную навеску воздушно-сухой небеленой целлюлозы разбивают в 270 см3 дистиллированной воды в гидрораэбивателе с частотой вращения 112..,118 с-1 до исчезновения комков, избегая разрезания волокон, Полученную массу переносят в реакционный стакан вместимостью 1 дм3. Сосуд, в котором разбивали массу, тщательно ополаскивают 100 см3 воды, которую также выливают в реакционный стакан. Пробу целлюлозы в стакане размешивают мешалкой до исчезновения комков массы. Не прерывая размешивания массы, приливают к ней смесь, состоящую из 50 см3 раствора перманганата калия концентрацией 0,1 моль/дм3 и 50 см3 раствора серной кислоты концентрацией 4 моль/дм3,- предварительно отмеренных пипетками в отдельный стакан. Одновременно с приливанием смеси в реакционный стакан включают секундомер. Стакан из-под смеси ополаскивают 30 см3 дистиллированной воды, которую также выливают в реакционный стакан, после чего общий объем жидкости в последнем должен составить 500 см3. Через 5 мин с момента приливания смеси замеряют температуру содержимого стакана. Реакция должна протекать при температуре °С. Точно через 10 мин от начала реакции в реакционный стакан добавляют мерным цилиндром 10 см3 раствора иодида калия концентрацией 1 моль/дм' для прекращения реакции окисления лигнина. Не прерывая размешивания, выделившийся свободный иод титруют раствором тиосульфата натрия концентрацией 0,2 моль/дм3 до соломенного цвета, затем добавляют 5... 10 капель 0,5%-ного раствора крахмала и продолжают титрование до обесцвечивания смеси.
Параллельно определяют расход тиосульфата натрия на титрование контрольной пробы. Для этого в реакционный стакан наливают 370 см3 дистиллированной воды и смесь, состоящую из 25 см3 раствора перманганата калия концентрацией 0,1 моль/дм3, 25 см3 дистиллированной воды и 50 см3 раствора серной кислоты концентрацией 4 моль/дм3, предварительно отмеренных пипеткой в другой стакан. Стакан из-под смеси ополаскивают 30 см3 дистиллированной воды, которую также выливают в реакционный стакан. Затем сразу же добавляют цилиндром 10 см3 раствора иодида калия концентрацией 1 моль/дм3 и при непрерывном перемешивании оттитровывают выделившийся иод раствором тиосульфата концентрацией 0,2 моль/дм3. Реакция должна протекать при температуре °С.
Б. При анализе полуцеллюлозы берут 100 см3 раствора перманганата калия концентрацией 0,1 моль/дм3, 100 см3 раствора серной кислоты концентрацией 4 моль/дм3 и 20 см3 раствора иодида калия концентрацией 1 моль/дм3. Общий объем должен составлять 1000 см3. Остальные условия анализа сохраняются неизменными.
При определении расхода раствора тиосульфата натрия на титрование контрольной пробы берут 50 см3 раствора перманганата калия концентрацией 0,1 моль/дм3, 50 см3 дистиллированной воды и 100 см3 раствора серной кислоты концентрацией 4 моль/дм3.
В. При анализе целлюлоз с небольшим содержанием лигнина подготовленную навеску разбивают в 100 см3 дистиллированной воды в гидроразбивателе до исчезновения комков, избегая разрезания волокон. Полученную массу переносят в реакционный стакан вместимостью 500 см1. Сосуд, в котором разбивали массу, тщательно ополаскивают є0 см3 дистиллированной воды, которую также выливают в реакционный стакан.
Пробу целлюлозы в реакционном стакане размешивают мешалкой до однородной массы. После этого, не прерывая размешивания, приливают к массе смесь, состоящую из 20 см3 раствора перманганата калия концентрацией 0,05 моль/дм3 и 20 см3 раствора серной кислоты концентрацией 4 моль/дм3, предварительно отмеренных пипетками в отдельный стакан, и одновременно включают секундомер. Стакан из-под смеси ополаскивают 30 см3 дистиллированной воды, которую также выливают в реакционный стакан. Общий объем жидкости в реакционном стакане должен составлять 200 см3. Через 5 мин с момента приливания смеси замеряют температуру реакции, которая должна протекать при °С.
Через 10 мин от начала реакции в реакционный стакан добавляют мерным цилиндром 5 см3 раствора иодида калия концентрацией 1 моль/дм3 для прекращения реакции окисления лигнина. Не прерывая перемешивания, выделившийся свободный иод титруют раствором тиосульфата натрия концентрацией 0,05 моль/дм3 в присутствии 0,5%-ного раствора крахмала до обесцвечивания смеси. Расход раствора перманганата калия на навеску целлюлозы должен составлять примерно % заданного на реакцию. Параллельно определяют расход раствора тиосульфата натрия на титрование контрольной пробы. Для этого в реакционный стакан наливают 130 см3 дистиллированной воды и смесь, состоящую из 10 см3 раствора перманганата калия концентрацией 0,05 моль/дм3, 10 см3 дистиллированной воды и 20 см3 раствора серной кислоты концентрацией 4 моль/дм3, предварительно отмеренных в стакан. Стакан из-под смеси ополаскивают 30 см3 дистиллированной воды, которую также выливают в реакционный стакан. Затем сразу добавляют 5 см3 раствора иодида калия концентрацией 1 моль/дм3 и при непрерывном перемешивании оттитровывают в присутствии крахмала выделившийся иод раствором тиосульфата натрия концентрацией 0,05 моль/дм3. Температура реакции при определении контрольной пробы должна быть °С.
А. Жесткость целлюлозы по перманганатному числу вычисляют по формуле

где d — коэффициент пересчета на 50%-ный расход перманганата калия, определяемый по табл. 3.3 в зависимости от значения и; т — масса абсолютно сухой целлюлозы, г; /0,1, где b — расход раствора тиосульфата натрия концентрацией 0,1 моль/дм3 на титрование контрольной пробы, см3; а — расход раствора тиосульфата натрия концентрацией 0,1 моль/дм3 на титрование рабочей пробы, см3; с — концентрация раствора тиосульфата натрия, моль/дм3.
6. Определение остаточного лигнина в технических целлюлозах УФ-спекрофотометрическим методом в кадоксене
Беленые и небеленые целлюлозы со сравнительно невысоким содержанием остаточного лигнина полностью растворяются в кадоксене и полученные растворы используют для УФ-спектрофотометрического определения лигнина. Измерение оптической плотности можно проводить только при длине волны около 280 им. Использовать более интенсивный максимум УФ-поглощения лигнина при длине волны 203...208 нм невозможно, так как кадоксен поглощает УФ-лучи в этой области, тогда как поглощение в области 280 нм незначительно. Экстрактивные вещества необходимо из исследуемой целлюлозы удалить предварительно. УФ-спектрофотометрический метод особенно ценен для беленых целлюлоз, для которых определение перманганатных чисел не пригодно, так как лигнин уже окислен отбеливающими реагентами. Этот метод позволяет определять остаточный лигнин в беленых целлюлозах при его массовой доле не менее 0,05%. Поскольку между концентрацией лигнина в кадоксеновом растворе и оптической плотностью последнего соблюдается прямолинейная зависимость, для расчета концентрации лигнина можно применять градуировочные графики или удельные коэффициенты поглощения. При малом содержании лигнина в беленых целлюлозах, когда оптическая плотность оказывается ниже 0,1, второй способ расчета более удобен.
Вследствие высокого рН кадоксена удельные коэффициенты поглощения хвойных лигнинов в кадоксене выше, чем в нейтральных или кислых растворах. В УФ-спектрах хвойных сульфатных лигнинов в кадоксене наблюдается плоский максимум при 300 нм, тогда как у лиственного лигнина в области 280...300 нм максимума практически нет и удельный коэффициент поглощения при длине волны 280 нм в отличие от диоксанлигнинов оказывается выше, чем у хвойного лигнина.
УФ-спектрофотометрический метод в определенной мере условен, так как остаточные лигнины в технических целлюлозах видоизменены по сравнению с природным лигнином и имеют отличающийся хромофорный состав. Единое мнение о наиболее подходящем препарате лигнина для построения градуировочного графика и определения удельного коэффициента поглощения отсутствует. Некоторые исследователи считают подходящим препаратом диоксанлигнин. В качестве препаратов для градуировки при анализе сульфатных целлюлоз наиболее целесообразно использовать сульфатные лигнины из черных щелоков от варки древесины хвойных и лиственных пород, а при анализе сульфитных целлюлоз — соответствующие лигносульфонаты. Несмотря на относительность метода, при малых содержаниях остаточного лигнина в целлюлозах УФ-спектрофотометрический метод дает более точные результаты, чем гравиметрический метод или метод определения жесткости по перманганатному числу.
Методика анализа. Навеску обессмоленной целлюлозы в виде воздушно-сухих отливок помещают в коническую колбу вместимостью 50 см3 с притертой пробкой. Влажность обессмоленной целлюлозы определяют в отдельной пробе. Добавляют отмеренные пипеткой 15 см3 кадоксена и встряхивают колбу в течение 2 ч. Если полностью прозрачный раствор не получается, то колбу с раствором оставляют на ночь в холодильнике при —15°С. После полного растворения содержимому колбы дают нагреться до комнатной температуры, добавляют пипеткой 15 см3 дистиллированной воды и встряхивают в течение 15 мин. Для получения более точных данных рекомендуется прозрачный при визуальном рассмотрении раствор подвергнуть центрифугированию. Затем заливают раствор в кювету толщиной 1 см и измеряют на УФ-спектрофотометре оптическую плотность при длине волны 280 нм, используя в качестве раствора сравнения смесь кадоксен-вода. Концентрацию раствора лигнина в кадоксен-воде, г/дм3, определяют по градуировочному графику или по рассчитанному с помощью графика удельному коэффициенту поглощения по формуле

где D — оптическая плотность при длине волны 280 нм
Массовую долю остаточного лигнина, % к исходной абсолютно сухой целлюлозе, рассчитывают по формуле

где с — концентрация лигнина в кадоксен-воде, г/дм'; g — масса абсолютно сухой навески обессмоленной целлюлозы, г; K>3> — коэффициент экстрагирования.
Построение градуировочиого графика. При анализе сульфатных целлюлоз в качестве эталонного препарата лигнина используют сульфатный лигнин, выделенный из черного щелока от варки целлюлозы соответственно из древесины хвойных или лиственных пород, например еловой или березовой. Готовят 10 растворов в кадоксен-воде в соответствии с методикой анализа с концентрацией эталонного лигнина от 0,005 мг/дм3 до 0,05 мг/дм3 и измеряют оптическую плотность, как указано выше. По полученным данным на миллиметровой бумаге строят график, откладывая по оси абсцисс концентрацию раствора лигнина в мг/дм3, а по оси ординат значения оптической плотности.
Примечание. При отсутствии препарата лигнина для построения градуировочиого графика для приближенного расчета можно использовать следующие удельные коэффициенты поглощения при длине волны 280 нм: для хвойного сульфатного лигнина 27,0 дм3 г' см; для лиственного сульфатного лигнина 30,2 дм3 г" см; для хвойных лигносульфонатов 11,0 дм3 -г-см-1.
7. Определение остаточных пентозанов
Для определения остаточных пентозанов в технических целлюлозах используют тот же метод, что и при анализе древесины. Он основан на образовании фурфурола из пентозанов при обработке целлюлозы 13%-ным раствором HCl при нагревании и последующем определении отогнанного фурфурола. Технические целлюлозы, особенно целлюлозы, полученные из древесины хвойных пород, содержат очень небольшое количество пентозанов по сравнению с древесиной. Побочные продукты, присутствующие в дистиллятах и определяемые вместе с фурфуролом гравиметрическим или титриметрическим методами, приводят к значительным ошибкам. Поэтому при анализе технической целлюлозы для определения фурфурола наиболее широко используются фотоколориметрические и спектрофотометрические методы. Фотоколориметрический метод основан на определении интенсивности окраски раствора фурфурола с орсином и позволяет получать более точные и воспроизводимые результаты. Спектрофотометрический метод дает возможность определять непосредственно фурфурол в дистилляте без использования цветных реакций.
При определении пентозанов в окисленных целлюлозах ошибка может возникать и в результате присутствия звеньев с карбоксильными группами у C>e>. Поэтому при расчете содержания пентозанов необходимо вносить поправку на фурфурол, образующийся из этих звеньев.
8. Определение пентозанов фотоколориметрическим методом
Методика анализа. Масса навески воздушно-сухой целлюлозы для определения пентозанов зависит от ожидаемой их массовой доли в испытуемой целлюлозе. При массовой доле пентозанов менее 0,4% масса навески составляет 5 г, от 4 до 2%—3 г и свыше 2% —1 г.
Навеску целлюлозы помещают в круглодонную колбу и туда же насыпают 20 г хлорида натрия. С помощью мерного цилиндра приливают в колбу 100 см3 13%-ного раствора HCl. Колбу помещают в баню с глицерином и присоединяют к установке, собранной в вытяжном шкафу. При этом уровень глицерина должен быть на 1,5...2,0 см выше уровня жидкости в колбе. Температуру глицериновой бани поддерживают в пределах 164...166°С и контролируют с помощью термометра, укрепленного в глицериновой бане. Отгонку дистиллята ведут в мерный цилиндр со скоростью 30 см3 за 10 мин. После отгонки каждых 30 см3 дистиллята из капельной воронки добавляют в колбу 30 см3 13%-ного раствора HCl. Конец отгонки фурфурола определяют по цветной реакции с анилинацетатом. Отогнанный дистиллят переносят в мерную колбу вместимостью 250 см3, цилиндр ополаскивают небольшим количеством 13%-ного раствора HCl и сливают в ту же колбу. Затем объем раствора в мерной колбе доводят до метки добавлением той же кислоты и содержимое колбы хорошо перемешивают. После этого определяют содержание фурфурола. Рекомендуется проводить анализ дистиллята сразу после перегонки, так как растворы фурфурола нельзя долго хранить. После перемешивания отбирают пипеткой 5 см3 раствора и переносят в коническую колбу с притертой пробкой вместимостью 100 см3. В колбу пипеткой вносят 25 см3 орсинового реагента, закрывают пробкой, содержимое колбы хорошо перемешивают и выдерживают в течение 50 мин при 0С. Затем в колбу приливают пипеткой 20 см3 этанола, снова перемешивают, слегка охлаждают и выдерживают в термостате при °С в течение 15 мин. Параллельную пробу готовят на 3...4 мин позднее первой. Таким же способом готовят контрольную пробу, в которой анализируемый раствор заменяют 5 см3 13%-ного раствора HCI. Выдерживают контрольную пробу вместе с испытуемыми пробами.
По истечении 15 мин на спектрофотометре при длине волны 630 нм или на фотоэлектрическом колориметре с- красным светофильтром определяют оптическую плотность испытуемых проб Относительно контрольной пробы. Применяют кюветы с толщиной поглощающего свет слоя 10, 20 или 50 мм. Предварительный выбор кювет производят таким образом, чтобы измеряемое значение оптической плотности находилось в пределах 0,3...0,5. По градуировочному графику находят массу пентозанов в испытуемой пробе целлюлозы в мг.
Массовую долю пентозанов, % к абсолютно сухой целлюлозе, вычисляют по формуле
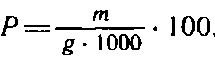
где т — масса пентозанов в испытуемой пробе целлюлозы, найденная по градуировочному графику, мг; g — масса абсолютно сухой навески целлюлозы, г.
Расхождение между результатами двух параллельных определений не должно превышать 0,2% при массовой доле пентозанов до 3% и 0,4% — при массовой доле их свыше 3%.
9. Определение пентозанов спектрофотометрическим методом
Фурфурол и гидроксиметилфурфурол, образующиеся из целлюлозы при нагревании с 12,,.13%-ным раствором HCI, дают сильную полосу поглощения в ультрафиолете. Максимумы поглощения находятся при длинах волн 277,5 и 284 нм соответственно, а коэффициенты поглощения при 277,5 нм и концентрации в мг/дм3 составляют 0,103 и 0,156.
Высокая чувствительность спектрофотометрического метода позволяет определить выход фурфурола из высокооблагороженных целлюлоз с низким содержанием пентозанов. Однако из таких целлюлоз образуется гидроксиметилфурфурола значительно больше, чем фурфурола, и вследствие подобия кривых поглощения этих двух компонентов становится невозможным их четкое различие.
Предложено несколько методов определения фурфурола в присутствии гидроксиметилфурфурола. Прежде всего выход фурфурола в отогнанном дистилляте может быть определен путем введения в расчетную формулу эмпирической поправки на гидроксиметилфурфурол. В основе другой методики находится разделение полученных альдегидов в делительной воронке между хлороформом, не содержащим этанола, и водной фазой. Измерение УФ-поглощения дистиллята и водной фазы позволяет рассчитать выход фурфурола.
Гидроксиметилфурфурол образуется примерно с одинаковой скоростью в течение всей перегонки. Если скорость перегонки сохраняется постоянной, концентрация гидроксиметилфурфурола в дистилляте после удаления всего фурфурола будет той же, что и в первой части, содержащей фурфурол. Для определения количества образующегося фурфурола измеряют УФ-поглощение дистиллята, используя в качестве раствора сравнения дополнительный дистиллят.
Определение фурфурола в присутствии гидроксиметилфурфурола производят также дифференциальным спектрофотометрическим методом на высокочувствительном УФ-спектрофотометре. При этом измеряют дифференциальные оптические плотности при 265 и 295 нм вместо максимумов индивидуальных соединений, а в качестве раствора сравнения используют раствор чистого фурфурола. Общая концентрация альдегидов должна быть около 10 г/дм3.
Концентрации фурфурола с>ф> и гидроксиметилфурфурола
с>гмф> в мг/дм3 рассчитывают по уравнениям
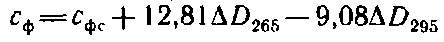 и
и
где с>фс> — концентрация раствора чистого фурфурола, мг/дм3; AD — дифференциальные оптические плотности при указанных длинах волн.
Массовую долю пентозанов, % к абсолютно сухой целлюлозе, вычисляют по формуле
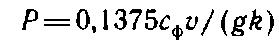 »
»
где х — объем дистиллята, см1; g — масса абсолютно сухой навески целлюлозы, г; k — коэффициент пересчета на пентозаны, равный 0,851.
Ниже приведены две методики определения низкого содержания пентозанов в целлюлозе спектрофотометрическнм методом.
Методика анализа с поправкой на гидроксиметилфурфурол.
Навеску воздушно-сухой целлюлозы массой от 1 до 5 г в зависимости от содержания пентозанов помещают в колбу и заливают 12%-ным раствором HCI. В отдельной пробе определяют влажность. Отгонку фурфурола ведут по методу TAPPI, но скорость перегонки уменьшают до 1,5 см3/мин и объем собираемого дистиллята до 200 см3. Для целлюлоз, содержащих 0,5...2% пентозанов, аликвотную пробу дистиллята н разбавляют водой в мерной колбе до 100 см3 и измеряют оптическую плотность при 277,5 нм, используя в качестве раствора сравнения дистиллированную воду. По градуировочному графику, построенному по чистому фурфуролу, находят его концентрацию в дистилляте. Массовую долю пентозанов, % к абсолютно сухой целлюлозе, рассчитывают по формуле

где g — масса навески абсолютно сухой целлюлозы, г; 1,563 — эмпирический коэффициент пересчета фурфурола в пентозаны; 0,002 — поправка на присутствие гидроксиметилфурфурола в дистилляте; т — общая масса фурфурола, определяемая по формуле т = с · 100 · 200/п, здесь с — концентрация фурфурола, найденная по градуировочному графику, мг/см3; х — объем аликвотной пробы, см3.
Методика анализа с получением дополнительного дистиллята. Навеску воздушно-сухой целлюлозы массой около 2 г помещают в колбу вместимостью 500 см3, заливают 100 см3 13,2%-ного раствора HCl и добавляют 20 г хлорида натрия. Перегонку ведут на устанозке со скоростью 2,5 см3/мин. По мере отгонки 25 см3 дистиллята из капельной воронки добавляют 25 см3 13,2%-ного раствора HCl. По достижении объема дистиллята 250 см3 в отдельную колбу собирают 20...25 см3 дополнительного дистиллята. Пипеткой отбирают по 10 см3 основного и дополнительного дистиллята, помещают их в мерные колбы вместимостью 1000 см3 и доводит
до метки дистиллирован ной водой. На УФ-спектрофотометре при длине волны 277 нм измеряют оптическую плотность, используя дополнительный дистиллят в качестве раствора сравнения. Оптическую плотность пересчитывают на фурфурол по градуировочному графику, построенному по данным для чистого фурфурола.
Массовую долю пентозанов, % к абсолютно сухой целлюлозе, рассчитывают по формуле
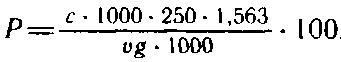
где с — концентрация фурфурола, определяемая по градуировочному графику, мг/см3; g — масса навески абсолютно сухой целлюлозы, г; х — объем аликвотной пробы, см3; 1,563 —эмпирический коэффициент пересчета фурфурола в пентозаны.
Построение градуировочиого графика. Готовят пять растворов чистого фурфурола с концентрацией 1, 2, 3, 4, 5 мг/см3 и измеряют их оптическую плотность на УФ-спектрофотометре при длине волны 277 нм в кювете толщиной 1 см. По полученным данным на миллиметровой бумаге строят градуировочный график, откладывая по оси абсцисс концентрацию раствора фурфурола в мг/см3, а по оси ординат значения оптической плотности.