Аналіз выкарыстання камп'ютэрў у біяінфарматыцы
ЗМЕСТ
ЗМЕСТ
УВОДЗІНЫ
ГЛАВА 1 АНАЛІЗ ВЫКАРЫСТАННЯ КАМП'ЮТЭРЎ У БІЯІНФАРМАТЫЦЫ
§1. Асноўныя прымяненні інфармацыйных тэхналогій
§2 Банкі даных біялагічнай інфармацыі
§3 Візуалізацыя біялагічнай інфармацыі
§4 Пашуковыя сістэмы
ГЛАВА 2 ВЫКАРЫСТАННЕ ІНФАРМАЦЫЙНЫХ ТЭХНАЛОГІЙ ДЛЯ ЗАДАЧ ПАРАЎНАННЯ Ў БІЯІНФАРМАТЫЦЫ
§1 Супастаўленне з узорам
§2 Параўнанне структур бялкоў
ГЛАВА 3
ВЫКАРЫСТАННЕ ПРАГРАМНЫХ БІБЛІЯТЭК
§1 Бібліятэка BioJava
Глава 4 ПРАКТЫЧНАЕ ВЫКАРЫСТАННЕ БІБЛІЯТЭКІ BIOJAVA
§1 Пастаноўка задачы
§2 Усталёўка бібліятэкі
§3 Выбар алгарытмаў
§4 Напісанне праграмы пошуку найбліжэйшых паслядоўнасцяў
§5 Тэсціраванне праграмы пошуку найбліжэйшых паслядоўнасцяў
ЗАКЛЮЧЭННЕ
СПІС ВЫКАРЫСТАНАЙ ЛІТАРАТУРЫ
ДАДАТАК 1
УВОДЗІНЫ
Сёння ўсё хутчэй нарастае аб'ём малекулярна-біялагічных і малекулярна-генетычных даных. Асэнсаванне гэтых каласальных па аб'ёмах даных ужо немагчыма без прыцягнення інфармацыйных тэхналогій і матэматыкі. Менавіта таму на стыку інфарматыкі, біялогіі і матэматыкі ўзнікла біяінфарматыка. Пад біяінфарматыкай зараз разумеюць прымяненне камп'ютэраў для вывучэння біялагічных аб'ектаў. У больш вузкім сэнсе яе можна разумець як прымяненне камп'ютэрных метадаў для вырашэння задач малекулярнай біялогіі. Сярод асноўных задач біяінфарматыкі вылучаюць:
вывучэнне геномаў
аналіз і прадказанне структуры бялкоў
аналіз і прадказанне ўзаемадзеяння бялкоў
Біяінфарматыка ўключае ў сябе наступныя элементы:
базы даных, у якіх захоўваецца інфармацыя пра біялагічныя аб'екты
набор інструментаў для аналізу і апрацоўкі гэтай інфармацыі
распрацоўку матэматычных мадэляў біялагічных аб'ектаў і алгарытмаў рашэння разнастайных біялагічных задач
Сярод банкаў даных найбольш вядомым прадстаўніком з'яўляецца Protein Data Bank (PDB) [1]. Гэта сховішча трохмернай структурнай інфармацыі аб бялках і нуклеінавых кіслотах. Гэтыя даныя звычайна атрымоўваюцца з дапамогай рэнтгенавай крысталаграфіі ці спектраскапіі на аснове ядзерна-магнітнага рэзанансу. Напаўняюць банк біёлагі і біяхімікі эксперыментальнымі данымі.
На бягучы час існуе шмат пакетаў праграм, якія прыназначаны для работы з біяфізічнай і біяхімічнай інфармацыяй. Вылучаюць наступныя накірункі:
візуалізатары інфармацыі з банкаў даных біялагічнай інфармацыі
прадказанне другаснай ці трэцічнай структуры бялку па вядомай першаснай структуры
прадказанне ўзаемадзеяння бялок-бялок і бялок-рэчыва
параўнанне бялкоў з банкаў даных і ацэнка іх падабенства
пабудова іерархічных дрэваў эвалюцыі біялагічных аб'ектаў
Варта заўважыць, што ўсе гэтыя банкі і праграмы пабудаваны з улікам існуючых алгарытмаў збору, захавання і апрацоўкі біялагічнай інфармацыі. З-за таго, што амаль усе задачы адносяцца да класу NP-поўных, то атрыманне рашэння ператвараецца ў складаную задачу. З-за медыцынскай важнасці гэтым праблемам прысвечана шмат увагі, што выклікае цеснае ўзаемадзеянне інфарматыкаў, біяхімікаў і біяфізікаў. Гэтыя праблемы і будуць асвечаны ў дадзенай працы.
ГЛАВА 1 АНАЛІЗ ВЫКАРЫСТАННЯ КАМП'ЮТЭРЎ У БІЯІНФАРМАТЫЦЫ
§1. Асноўныя прымяненні інфармацыйных тэхналогій
Як ужо было сказана, выкарыстанне камп'ютэраў для вырашэння біялагічных задач стала неабходным. Асноўныя сферы іх прымянення наступныя:
камунікацыя
кіраванне прыстасаваннямі
вылічэнні
захаванне
Камп'ютэры шырока выкарыстоўваюцца для кіравання разнастайнымі прыстасаваннямі для працы з біялагічнымі аб'ектамі: машынамі для секвенціравання ДНК, брадзільнымі чанамі, біярэактарамі і гэтак далей. Гэтыя устаноўкі кіруюцца праграмуемымі рукамі-робатамі і змяншаюць патрэбу ў людзях-аператарах. Там, дзе вельмі важны час выканання, камп'ютэры выйграюць у людзей, бо могуць працаваць без перапынкаў і з вялікай дакладнасцю.
З развіццём сеткавых тэхналогій і персанальных камп'ютэраў падрыхтоўка вучоным артыкула стала не толькі ў разы хутчэйшым, але і яго распаўсюджванне таксама паскорылася. Зараз мала хто з вучоных будзе адразу звяртацца да папяровага выдання, а хутчэй за ўсё звернецца да адной са шматлікіх баз даных біялагічных структур. Стала магчымай сумесная праца вучоных у геаграфічна аддаленых рэгіёнах.
Вылічальныя задачы ў біяінфарматыцы прадстаўлены вельмі шырока: ад візуалізацыі і пошуку падобных амінакіслотных ланцужкоў да мадэліравання ўзаемадзеяння складаных бялкоў па трохмерных іх структурах. Рашэнню такіх задач дапамагае вялікая колькасць гатовых праграмных сродкаў, а таксама наяўасць гатовых праграмных бібліятэк, якія спрашчаюць напісанне новых праграм і падтрымку старых.
Функцыі захавання шырока прадстаўлены ў выглядзе разнастайных сховішчаў інфармацыі, адрозных па сваёй эфектыўнасці, даступнасці і характары інфармацыі, для працы з якой яны прыназначаны.
§2 Банкі даных біялагічнай інфармацыі
Адной з найважнейшых частак прымянення камп’ютэраў у малекулярнай біялогіі з’яўляецца захаванне біялагічнай інфармацыі. Для гэтай мэты існуе шэраг спецыяльных сховішчаў, гэтак званых банкаў даных ці баз даных. Яны знаходзяцца ў цэнтры выкарыстання інфармацыйных тэхналогій у біяінфарматыцы. Насамрэч, большасць аперацый па вылічэннях і камунікацыі заснавана на даных, што знаходзяцца ў банках даных. Інфармацыя ў іх захоўваецца на трывалай і доўгачасовай аснове, амаль не падвяргаецца зменам, і яе форма прыстасаваная для хуткага выканання аперацый пошуку, сартыроўкі і іншых.
Існуе шэраг розных тыпаў такіх сховішчаў: банкі нуклеатыдных паслядоўнасцяў, бялковых паслядоўнасцяў, трохмерных структур, паслядоўнасных матываў (матыў – невялікі структурны элементы, які сустракаецца ў многіх пратэінах, напрыклад, α-спіраль). У табліцы 1.1 прыведзены некаторыя прадстаўнікі кожнага тыпу баз даных [2].
Табліца 1.1. Тыпы баз біялагічных даных
|
Тып базы даных |
Прыклады |
Заўвага |
|
Нуклеатыдныя паслядоўнасці |
GenBank |
Адна з найвялікшых баз даных паслядоўнасцяў |
|
EMBL |
Еўрапейская лабараторыя малекулярнай біялогіі |
|
|
NDB |
База даных нуклеінавых кіслотаў |
|
|
Бялковыя паслядоўнасці |
SWISS-PROT |
Швейцарскі і Еўрапейскі інстытуты біяінфарматыкі |
|
PIR |
Рэсурс інфармацыі пра бялкі |
|
|
Трохмерныя структуры |
PDB |
Банк пратэінавых даных |
|
MMDB |
База даных малекулярнага мадэліравання |
|
|
Паслядоўнасныя матывы |
LIGAND |
Хімічныя структуры і рэакцыі |
|
PROSITE |
Паслядоўнасныя матывы |
|
|
ProDOM |
Бялковыя дамены |
Акрамя публічных, ёсць шэраг прыватных базаў даных, які хутка пашыраецца. У яго ўваходзяць базы, стварэннем і падтрымкай якіх займаюцца кампаніі і камерцыйныя лабараторыі пры падтрымцы акадэмічных інстытутаў. Напрыклад, база даных LifeSeq ад кампаніі Incyte Genomics, Inc. утрымоўвае паслядоўнасці генаў ад людзей, пацукоў і мышэй.
Незалежна ад таго, прыватнай ці публічнай з’яўляецца такая база, усе яны выконваюць адмысловыя функцыі па працы з біялагічнай інфармацыяй (пераважна функцыі пошуку, захавання і накаплення).
§3 Візуалізацыя біялагічнай інфармацыі
Бялок — гэта складаная малекула, для якой яе лінейная структура, зададзеная паслядоўнасцю амінакіслот, вызначае унікальную трохмерную фігуру. Гэтая фігура з'яўляецца адной з найважнейшых характарыстык бялка, бо цалкам адказвае за функцыі бялка і яго ўзаемадзеянне іншымі малекуламі. Гэтае ўзаемадзеянне часта заснавана на дапасаванасці фігур, гэта значыць, што ў бялкоў ёсць выпукласці і вогнутасці, якія дазваляюць ім звязвацца адзін з адным і ўтвараць складаныя структуры, такія як скура ці валасы. З гэтае прычыны такую важную ролю пры вынаходніцтве лекаў адыграе пошук малекул, чыя трохмерная форма дазволіць ім далучацца да зададзеных бялкоў ці ферментаў з мэтай замарудзіць ці паскорыць іх дзеянне.
Падчас эвалюцыі ў чалавека добра развілася засвойванне графічнай інфармацыі і распазнаванне вобразаў. Табліцы, дыяграмы і малюнкі часта суправаджаюць многія працы для прасцейшага ўсведамлення інфармацыі. У біяінфарматцы разнастайныя звесткі прадстаўлены ў абстрактнай форме, якая патрабуе далейшай візуалізацыі. Гэта асабліва запатрабавана для візуалізацыі паслядоўнасцяў і структур бялкоў, а таксама для стварэння графічных інтэрфейсаў для карыстальнікаў. Яшчэ адным накірункам з’яўляецца дапамога лікаваму аналізу, асабліва статыстычнаму. У кожнай вобласці прымянення замена тэкставай і лікавай інфармацыі графічнай мае мэтай замену чытання і далейшай лагічнай і матэматычнай інтэрпрэтацыі больш хуткім распазнаваннем вобразаў.
Асноўнай задачай тут з’яўляецца візуалізацыя трохмерных структур бялкоў, з мэтай пошуку лекаў. Візуалізацыя першаснай структуры бялка дае мала ведаў пра яго функцыі. У адрозненне ад візуалізацыі структур вышэйшых парадкаў. Так, разуменню докінгу бялкоў і параўнанню іх структур вельмі спрыяюць трохмерныя выявы гэтых малекул.
Існуе шэраг праглядальнікаў файлаў з такіх банкаў, якія канструіруюць трохмерную выяву бялка. Яны ўсе адрозніваюцца па сваіх магчымасцях і характарыстыках. Пры выбары канкрэтнага сродка трэба кіравацца прастатой выкарыстання, хуткасцю праграмы, апаратнымі патрабаваннямі, дакументацыяй і падтрымкай, а таксама коштам.
Адной з такіх праграм іх з'яўляецца Accelrys.[3] З яе дапамогай даследчык можа візуальна ацаніць структуру бялка. Праграма мае шэраг розных опцый і наладак, магчымасць маштабіравання і павароту выявы, уключэнне і выключэнне паказу зададзеных структурных элементаў, з'яўляецца бясплатнай і кросплатформавай. Наяўнасць такіх візуальных сродкаў дапамагае даследчыку ўбачыць структуру бялка і, напрыклад, параўнаць яе са структурай іншага бялка. На малюнку 1.1 прадстаўлена адна з магчымых форм візуалізацыі бялку.
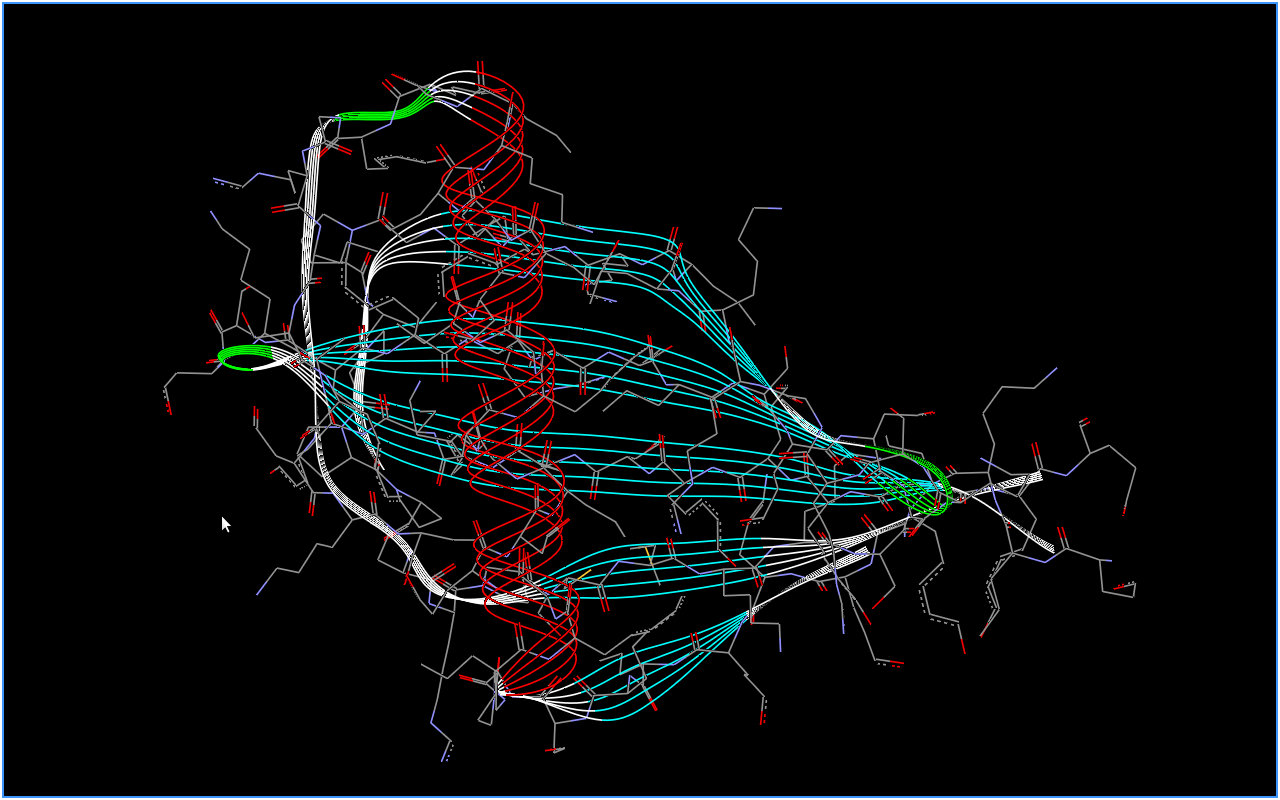
Малюнак 1.1 - Прадстаўленне трохмернай структуры бялка з дапамогай Accelrys
Акрамя гэтага ёсць праглядальнікі інфармацыі з генетычных баз даных. Прыкладам такой праграмы з’яўляецца NCBI’s Web-base Map Viewer[4]. Гэта анлайнавая праграма з веб-інтэрфейсам, якая праводзіць пошук па некалькіх анлайнавых базах NCBI. Яна дазваляе вучонаму вызначаць месцазнаходжанне канкрэтнага гена ў геноме арганізма, адлегласць паміж генамі ў геноме і прадастаўляе даныя пра ген у нейкім вызначанай храмасомнай вобласці. Праграма працуе ў браўзеры, а ўсе вылічэнні праводзяцца на баку сервераў NCBI. У адрозненне ад аналізу і пабудовы трохмерных структур бялкоў, вылічэнні для візуалізацыі аднамернай паслядоўнасці невялікія.
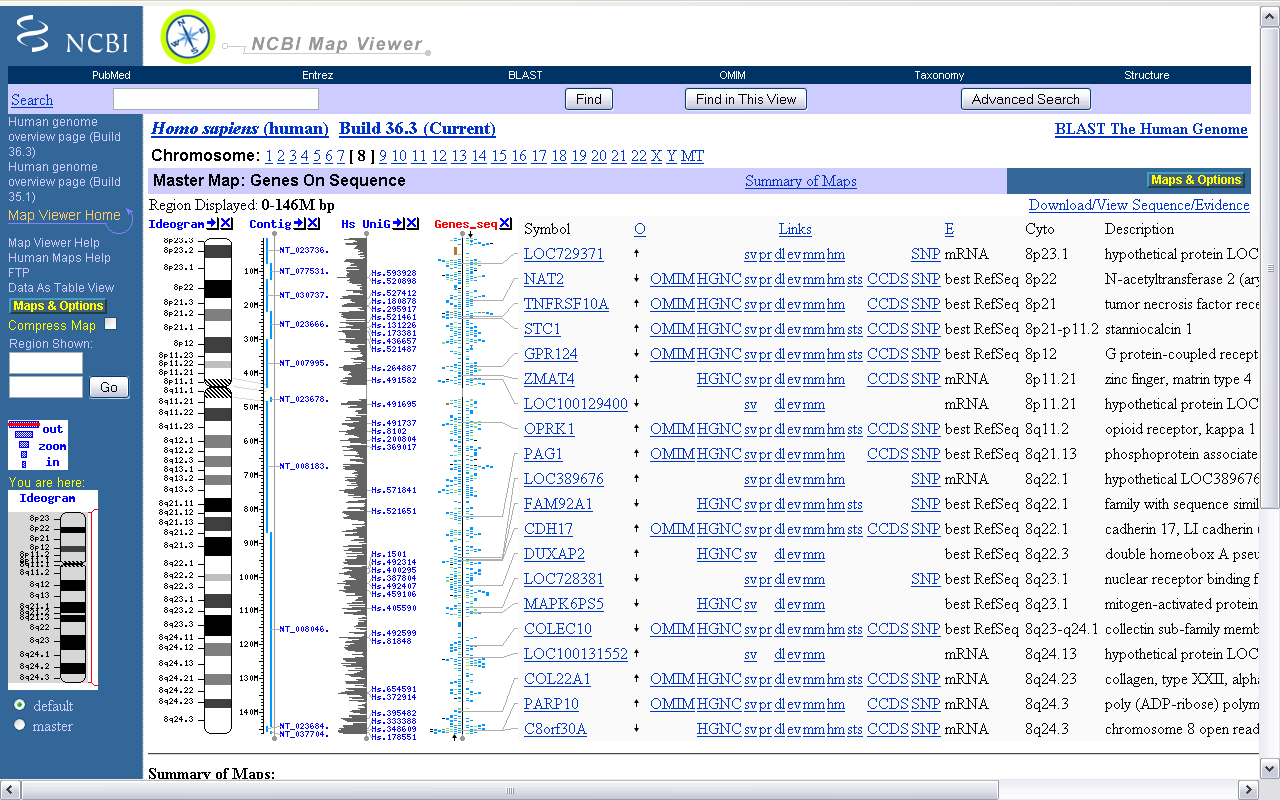
Малюнак 1.2. Даныя пра 8-ю храмасому Homo Sapiens у NCBI’s Map Viewer
На малюнку 1.2 прадстаўлены даныя, атрыманыя з дапамогай NCBI’s Map Viewer, пра восьмую храмасому Homo Sapiens.
§4 Пашуковыя сістэмы
Адной з мэтаў развіцця біяінфарматыкі з’яўляецца аўтаматызацыя дыягностыкі хвароб і лячэння. Калі кансультацыю з канкрэтным доктарам заменіць канферэнцыя з камп’ютэрам, і пры адсылцы нейкага ўзору, напрыклад, сліны ці крыві, камп’ютэр мог бы прааналізаваць яго, паставіць дыягназ на аснове інфармацыі са шматлікіх баз даных (генетычны профіль пацыента і сям’і, звычкі і абставіны працы, гісторыя захворванняў і іншыя) і прызначыць лячэнне, якое будзе найлепш падыходзіць.
Аднак такі падыход патрабуе сістэмы сувязяў паміж разнастайнымі базамі біялагічных і медыцынскіх даных. Напрыклад, у генетычным профілі павінны прысутнічаць спасылкі на нейкія вобласці ў базах даных нуклеатыдных паслядоўнасцяў, бялковых паслядоўнасцяў, ферментаў і схільнасцямі да хвароб. Такія сувязі неабавязкова відавочныя ці, нават, вядомыя зараз.
На сённяшні дзень гэтыя сувязі паміж данымі ў лічбавым фармаце недастаткова гатовыя, бо знаходзяцца ў розных базах даных. Амаль усе яны пабудаваны на аснове розных і несумяшчальных тэхналогій і выкарыстоўваюць розныя слоўнікі і мовы для аперацый з данымі. Гэта выклікана тым, што базы ствараюцца як асобныя праграмы для выканання некалькіх функцый, а пра іх звязванне задумваюцца звычайна пасля стварэння. Сувязі паміж базамі могуць стварацца дынамічна з дапамогай пашуковых сістэм.
Пры пошуку рашэння задачы малекулярнай біялогіі часта неабходна пераходзіць ад адной базы да іншай і вяртацца назад. Каб дапамагчы ў гэтым працоўным працэсе былі створаны сістэмы атрымання інфармацыі, з зададзеным загадзя сувязямі паміж элементамі ключавых анлайнавых баз даных. Найбольш вядомай з такім інтэгратыўных сістэм з’яўляецца Entrez Нацыянальга Цэнтра Біятэхналагічнай Інфармацыі (NCBI)[5]. Асноўныя базы, якія ўваходзяць у NCBI прадастаўлены на табліцы 1.2.[2]
Табліца 1.2 Базы даных у сістэме Entrez
|
База даных |
Апісанне |
|
PubMed |
Біямедычная літаратура |
|
Бялкі |
Бялковыя паслядоўнасці з PIR, SWISS-PROT, PDB, EMBL |
|
Нуклеатыды |
Нуклеатыдныя паслядоўнасці з GenBank, EMBL, DDJB (японская база даных ДНК) |
|
Структуры |
Трохмерныя структуры з PDB |
|
Геномы |
Разнастайныя базы з генетычнай інфармацыяй |
|
OMIM |
Чалавечыя гены і генетычныя захворванні |
|
Таксаномія |
Іерархія відаў арганізмаў у генетычнай базе даных NCBI |
|
Трохмерныя дамены |
Дамены бялкоў з базы даных NCBI закансервіраваных даменаў |
У гэтай сістэме сувязі не толькі паміжбазавыя, але і ўнутраныя. Напрыклад, не толькі сувязі прысутнічаюць не толькі паміж PubMed і базай нуклеатыдаў, але і паміж падобнымі элементамі ў самой базе нуклеатыдаў.
Ёсць дзве версіі сістэмы Entrez: адна, якая выкарыстоўвае праграму, што выконваецца лакальна на працоўнай станцыі карыстальніка, называецца Networked Entrez, а другая даступная праз веб-браўзер. Networked Entrez узаемадзейнічае напрамую з дыспетчэрам NCBI з дапамогай усталяванага злучэння кліент-сервер. Аднак, з-за таго, што Network Entrez можа выкарыстоўваць лакальныя вылічальныя рэсурсы, яна можа працаваць значна хутчэй за браўзерны варыянт. Да таго ж яна прадастаўляе больш багаты і зручны інтэрфейс з некалькімі ўкладзенымі вокнамі і больш зручнымі праглядальнікамі паслядоўнасцяў геномаў і трохмерных структур бялкоў. Аднак перавагамі браўзернай версіі з’яўляецца адстутнасць неабходнасці абнаўляць праграму, усталёўваць і наладжваць яе. Таксама браўзерная версія прадстаўляе больш зручны інтэрфейс для пераходу па гіпертэкставых спасылках на старонках адлюстравання інфармацыі.
ГЛАВА 2 ВЫКАРЫСТАННЕ ІНФАРМАЦЫЙНЫХ ТЭХНАЛОГІЙ ДЛЯ ЗАДАЧ ПАРАЎНАННЯ Ў БІЯІНФАРМАТЫЦЫ
§1 Супастаўленне з узорам
Аўтаматычнае супастаўленне з узорам – гэта магчымасць праграмы параўноўваць невядомыя і вядомыя ўзоры і вызначаць іх ступень падабенства. Складанасць такой задачы заключаецца ў тым, што трэба не проста знайсці адпаведнік дадзенаму ўзору, а хутка знайсці адзін ці некалькі адпаведнікаў з вялікай базы даных на рэсурсах, якія ёсць у распараджэнні. У дадатак да гэтага, часта паўстае задача знайсці адпаведнікі, якія амаль адпавядаюць ці з’яўляюцца падобнымі да дадзенага ўзора, але паняцце падабенства не вельмі строга азначана ў праграмным ці біялагічным сэнсах.
Выраўніванне паслядоўнасцяў – працэс супастаўлення іх адна пад адной так, каб колькасць супадзенняў была максімальнай, пры гэтым за пропускі і несупадаючыя сімвалы бяруцца нейкія штрафы. Выраўніванне – гэта фундаментальны сродак для вызначэння гамалагічнасці (наяўнасці агульнага продка) і функцыяльнасці бялкоў. Так, калі частка адной паслядоўнасці супадае з часткай другой паслядоўнасці, то можна казаць аб важнасці гэтага кавалка ў эвалюцыйным адборы і рабіць нейкія высновы пра яго функцыянальнасць.
Адрозніваюць некалькі тыпаў выраўніванняў:
папарнае
глабальнае і лакальнае
множнае
Для папарнага выраўнівання характэрна супастаўленне двух паслядоўнасцяў. Няхай у нас ёсць дзве паслядоўнасці ATTCAGTGCT і ATTGCT. І трэба іх выраўняць. Ацэнка падабенства будзе вылічвацца як:
колькасць супаўшых – каэфіцыент разрыву * колькасць разрываў – каэфіцыент несупадзенн * колькасць несупадзенняў.
Для гэтага прыкладу пры адзінкавых каэфіцыентах найлепшым будзе наступнае выраўніванне з велічынёй падабенства 2:
ATTCAGTGCT
ATT----GCT
Адрозненне глабальнага ад лакальнага выраўніванняў заключаецца ў тым, што ў глабальным мы максімізуем функцыю падабенства па ўсёй даўжыні паслядоўнасці, а ў лакальным – кавалачна. Глабальнае выраўніванне дасць нам прадстаўленне аб падабенстве бялкоў і, напрыклад, пра адносіны іх да аднаго сямейства. Лакальнае ж можа сведчыць пра аднолькавыя функцыі, якія выконваюць падобныя кавалкі. Для вырашэння задачы лакальнага выраўнівання ёсць некалькі алгарытмаў, якія адрозніваюцца сваімі характарыстыкамі. Адным з іх з’яўляецца алгарытм Сміта-Уотэрмана.
Задача множнага выраўнівання заключаецца ў тым, што ў нас адначасова выраўніваецца больш за дзве паслядоўнасці. Гэтая задача нашмат больш складаная за папарнае выраўніванне. Але і для яе таксама ёсць рашэнні. Зразумела, што пры дастаткова вялікай даўжыні паслядоўнасці, задача не можа быць вырашана ручнымі метадамі. Таму стала неабходна выкарыстанне апарату тэорыі алгарытмаў і вылічальных машын.
Найбольш папулярнымі метадамі вырашэння задачы выраўнівання з’яўляюцца: баесаўскія метады, дынамічнае праграміраванне, нейронныя сеткі, генетычныя алгарытмы, тэхнікі, заснаваныя на словах, ацэначныя матрыцы. Сярод іх найбольш часта выкарыстоўваюцца методыкі дынамічнага праграміравання і тэхнікі, заснаваныя на словах.
З-за відавочнай важнасці рашэння такой задачы быў створаны шэраг праграмных сродкаў. Выдзяляюцца сродкі для выраўнівання нуклеатыдных і бялковых паслядоўнасцяў. Да нуклеатыдных адносяцца: BLASTN, BLASTX, BALSA. Да бялковых адносяцца: BLASTP, Smith-Waterman, PHI-BLAST.
Найбольш вядомым і выкарыстоўваемым сярод праграм для выраўнівання нуклеатыдаў з’ўяляецца BLASTN [6] і яго вытворныя. Праграма даступная праз веб-інтэрфейс. На малюнку 2.1 паказана працоўная вобласць запытаў да гэтатай праграмы. Праграма з падобным інтэрфейсам BLASTP існуе і для пошуку і параўнання бялок-бялок.
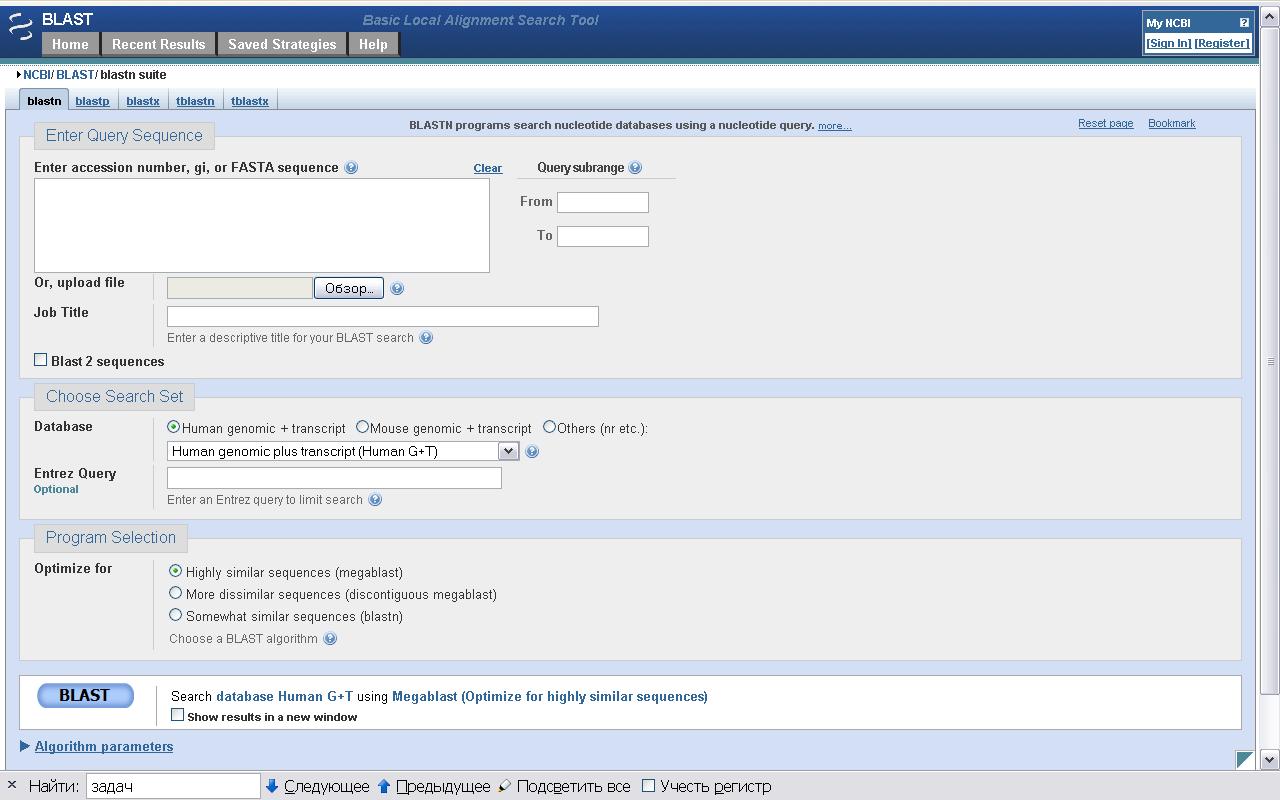
Малюнак 2.1, Працоўная вобласць BLASTN
§2 Параўнанне структур бялкоў
За апошняе дзесяцігоддзе было распрацавана шмат алгарытмаў і праграмных сродкаў для апрацоўкі біялагічных бялковых даных. Сярод іх вылучаюць два галоўных накірункі: прадказанне структуры бялка і параўнанне структур бялкоў. Першы звязаны з вызначэннем другаснай ці трэцічнай структуры па паслядоўнасці амінакіслот. А другі звязаны з параўнаннем атрыманых эксперыментальна ці тэарэтычна структур біялагічных аб'ектаў. Гэтае параўнанне можа быць выкарыстана для вызначэння функцый новых бялкоў на аснове падабенства з ужо даследаванымі, групіроўкі іх у сямействы, а таксама для ацэнкі прадказаных структур, пры параўнанні іх са структурамі атрыманымі эксперыментальна.
У аснове гэтага праграмнага забеспячэння ляжыць інфармацыя пра структуры бялкоў, якая захоўваецца ў спецыяльных базах даных. Існуе некалькі сховішчаў такой інфармацыі. Найбуйнейшым з іх з'яўляецца PDB. Зараз у ім сабраная інфармацыя больш чым на 50000 бялкоў. Захоўваецца яна ў тэкставых файлах спецыяльнага фармату і знаходзіцца ў свабодным доступе. На малюнку 2.2 паказана сціслае апісанне бялка-звязкі пеніцыліну 1TVF з сайта PDB. Акрамя PDB існуе база даных SCOP (структурная класіфікацыя бялкоў), усе бялкі ў якой звязаныя паміж сабой адносінамі падабенства ці эвалюцыйнымі. Бялкі аб'ядноўноўваюцца ў сямействы і суперсямействы. Таму гэтая база прадастаўляе хуткі пошук бялкоў, блізкіх да зададзенага.
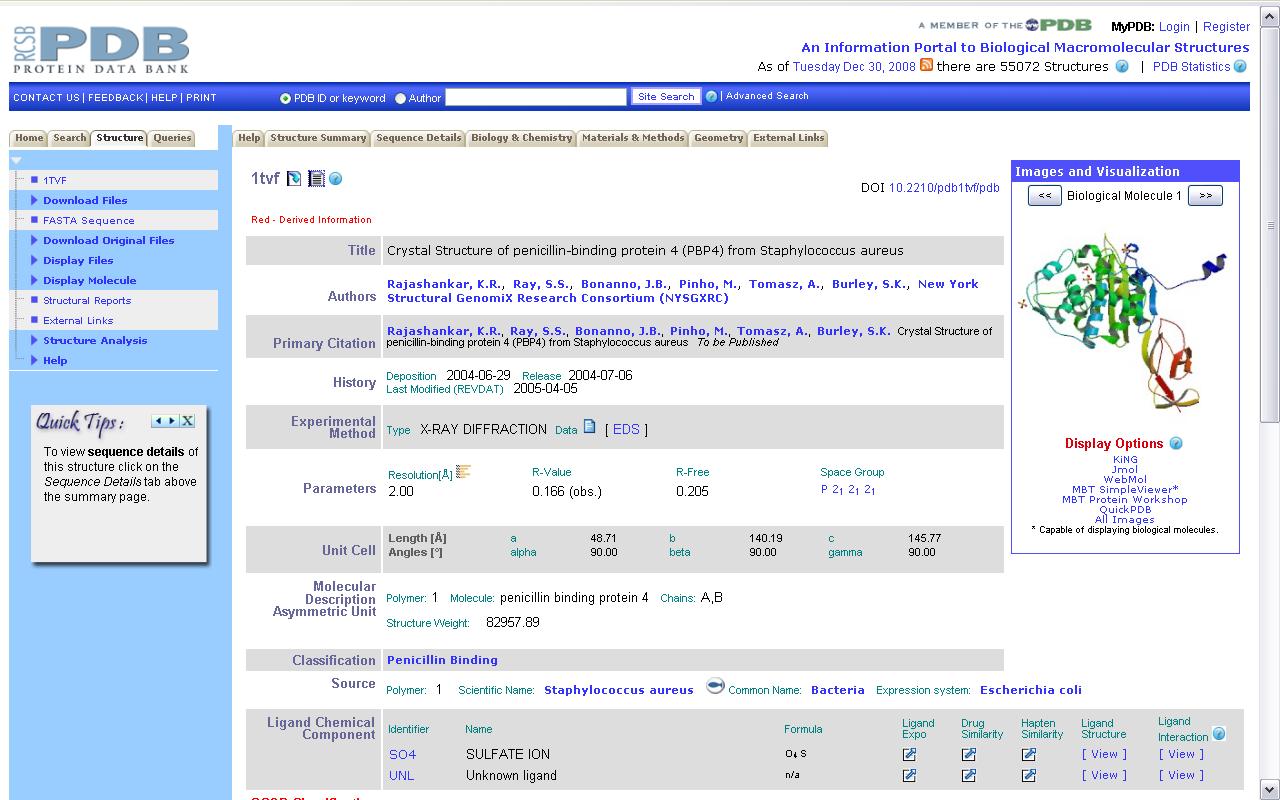
Малюнак 2.2. Бялок-звязка для пеніцыліну
Задача параўнання бялкоў па іх структурах з'яўляецца ў агульным выпадку NP-поўнай. Таму нядзіўна, што акрамя дакладных алгарытмаў пры ўваходных даных з нейкімі абмежаваннямі, існуе цэлы шэраг прыблізных алгарытмаў, заснаваных на розных эўрыстычных падыходах.
Найбольш вядомымі з'яўляюцца алгарытмы Maxsub>, GDT, 3dSearch, TM-align, DALI. Агульным для большасці алгарытмаў у гэтай вобласці з'яўляецца тое, што яны не з'яўляюцца прыблізнымі (гэта значыць не даюць рашэнне, якое ляжыць у гарантаванай блізкасці ад дакладнага рашэння), а з'яўляюцца эўрыстыкамі, якія заснаваны на нейкіх рацыянальных ідэях, што змяншаюць прастору пошуку рашэння.
На аснове гэтых алгарытмаў з’яўвіўся шэраг праграм, якія дапамагаюць біяёлагам параўноўваць трохмерныя структуры не толькі інтуітыўна, а з дапамогай вылічальных машын. Адным з прадстаўнікоў такога праграмнага забеспячэння з’яўляецца сервіс 3-Dimesional Structural Superposition (3DSS)[7], які прадстаўляе праз веб-інтэрфейс функцыянальнасць для параўнання бялкоў з PDB. Інтэрфейс рэсурса нескладаны і ўся аперацыя займае некалькі крокаў. Спачатку трэба выбраць якія бялкі мы параўноўваем. Для гэтага неабходна ўвесці ідэнтыфікатары бялкоў з PDB ці загрузіць любы файл у PDB-фармаце. Потым выбраць часткі бялка для параўнання і параметры суперпазіцыі. І, нарэшце, ў браўзеры можна будзе ўбачыць трохмерную выяву, дзе будзе зроблена суперпазіцыя бялкоў і іх можна адрозніць па колеры. Вынік працы праграмы можна ўбачыць на малюнку 2.3 (параўноўваліся бялкі 1SDB i 1HOE).
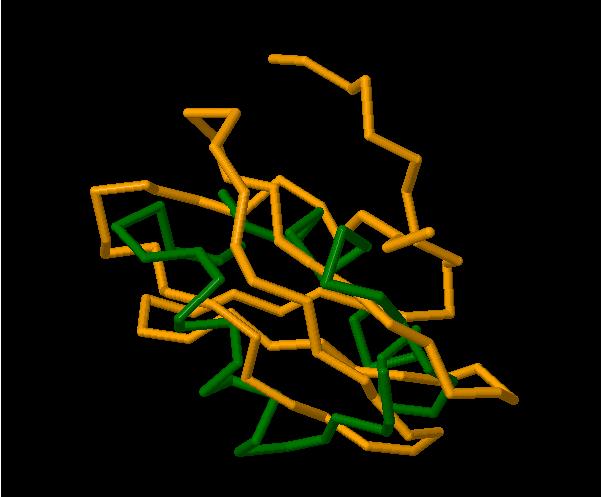
Малюнак 2.3. Суперпазіцыя бялкоў у 3DSS
Яшчэ адным вядомым анлайнавым сервісам, які прадастаўляе паслугі па супастаўленні трэцічных структур бялкоў, з’ўялецца MATRAS. Ён уключае ў сябе набор праграм для структурнага параўнання. Туды ўваходзяць праграмы для папарнага параўнання бялкоў, множнага параўнання, знаходжання падобных участкаў на адным бялку, параўнанне бялка з цэлай бібліятэкай бялкоў, а таксама пошук паслядоўнасці амінакіслотаў у PDB. Інтэрфейс таксама дастаткова просты і дазваляе хутка параўнаць бялкі і атрымаць шмат карыснай інфармацыі пра іх падабенства, не толькі візуальнай, але і лікавай. Таксама прадастаўлена магчымасць візуалізацыі з дапамогай разнастайных плагінаў, напрыклад, з дапамогай Java аплета Jmol. Вывад тэкставай інфармацыі прадастаўлены на малюнку 2.4 (параўноўваліся бялкі з ідэнтыфікатарамі 1MDB i 4HHB)
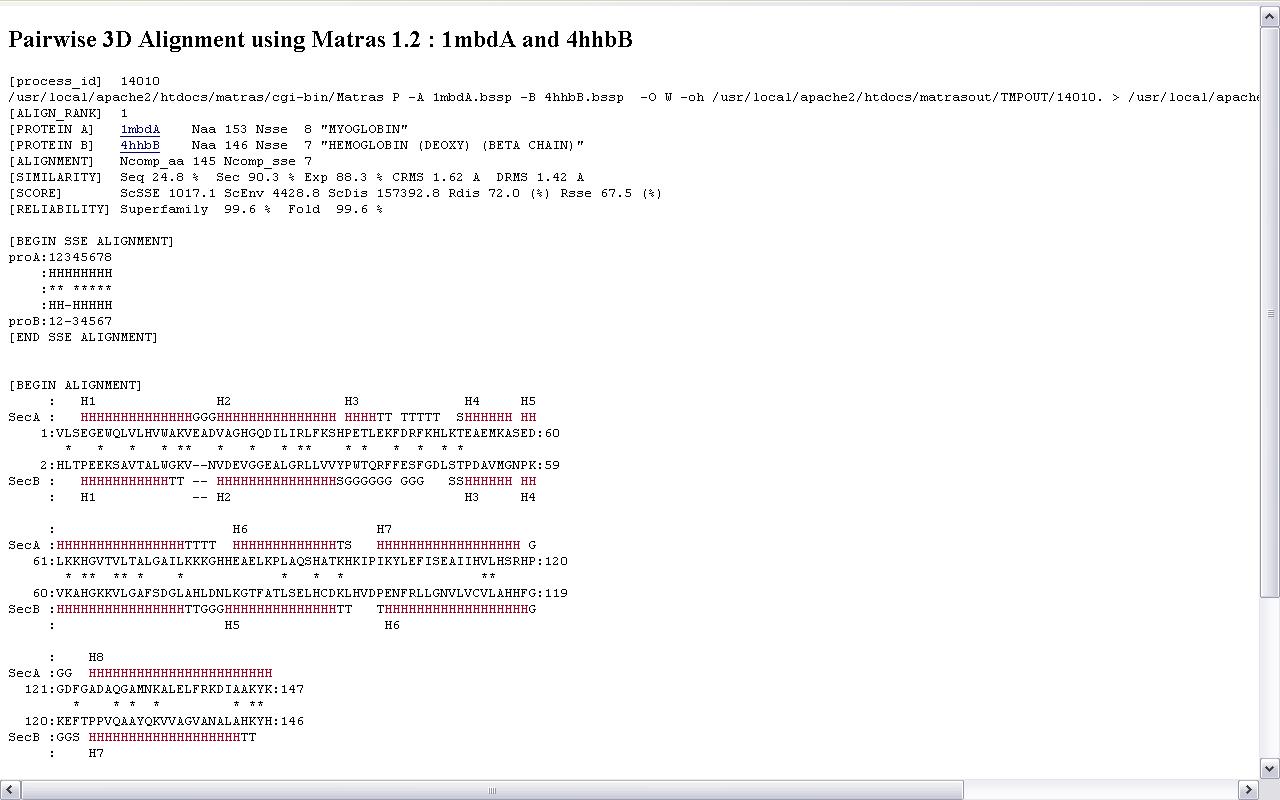
Малюнак 2.4. Вынік працы MATRAS
ГЛАВА 3 ВЫКАРЫСТАННЕ ПРАГРАМНЫХ БІБЛІЯТЭК
Раней былі апісаныя многія гатовыя праграмныя сродкі, якія можна было выкарыстоўваць для выканання нейкіх задач біяінфарматыкі. Але для гэтага неабходна шукаць гатовае рашэнне сярод існуючых праграмных сродкаў і рабіць правільны выбар іх камбінацыі. Таму можа ўзнікнуць праблема таго, што гатовага рашэння можа і не быць у дадзены момант. Дапамагчы ў гэтай сітуацыі могуць гатовыя прыкладныя праграмныя інтэрфейсы (ППІ) для розных моў праграмавання. Яны дазволяць у сціслы і хуткі тэрмін напісаць даследчыку праграму на аснове гатовай бібліятэкі класаў і функцый вырашыць канкрэтную задачу без прыцягнення іншых распрацоўчыкаў. Асаблівую вядомасць зараз маюць праекты BioJava [2] і BioPerl.
§1 Бібліятэка BioJava
Праект BioJava быў распачаты ў 1999 годзе Томасам Даўнам і Мэцью Покакам як ППІ для спрашчэння напісання праграм для біяінфарматыкі з выкарыстаннем мовы праграмавання Java. Цяпер гэта паўнавартасная аснова для стварэння праграм на Java і выканання многіх распаўсюджаных заданняў біяінфарматыкі. Гэтая бібліятэка адносіцца да праектаў з адкрытым праграмным кодам. Яна распаўсюджваецца па ліцэнзіі LGPL, таму можа быць выкарыстана і распаўсюджана ў любой форме[2]. Сярод магчымасцяў, якія прадастаўляе гэта бібліятэка вылучаюць 10 галоўных:
Нуклеатыдныя і амінакіслотныя алфавіты
Разборшчык BLAST
Увод/вывад паслядоўнасцяў
Дынамічнае праграміраванне
Увод/вывад структур і маніпуляцыі з імі
Маніпуляцыі з паслядоўнасцямі
Генетычныя алгарытмы
Статыстычыя размеркаванні
Графічныя інтэрфейсы карыстальніка
Захаванне ў базе даных
Для працы з апошнімі версіямі бібліятэкі неабходны віртуальная машына Java 1.5 і шэраг бясплатных бібліятэк, на якія ёсць залежнасці ў BioJava. Разам з бібліятэкай ідзе падрабязная і змястоўная дакументацыя і прыклады, якія дапамогуць разабрацца ў кароткі тэрмін з асноўнымі магчымасцямі ППІ. Разгледзім некаторыя гэтыя магчымасці больш падрабязна.
Разнастайныя біялагічныя аб’екты апісваюцца рознымі спосабамі. Звычайна гэта робіцца на аснове запісу іх дробных элементарных аб’ектаў нейкімі сімваламі. Сімвалы могуць брацца з разнастайных алфавітаў, спецыфічных да канкрэтнай вобласці біяінфарматыкі. Гэтак, пры апісанні паслядоўнасцяў ДНК нам неабходны алфавіт усяго з чатырох сімвалаў тымін (Т), гуамін (Г), цытазін (Ц) і адэнін (А). Для апісання поліпептыдаў патрэбны ўжо алфавіт з 20 сімвалаў (бо ў поліпептыды ўваходзяць 20 розных амінакіслотных рэшткаў). Такім чынам, для абстракцыі працы з разнастайнымі паслядоўнасцямі сімвалаў і алфавітамі і была ўведзена іерархія тыпаў для розных структур. Гэтая функцыянальнасць размешчана ў пакетах org.biojava.bio.symbol і org.biojava.bio.seq.
Для спрашчэння працы са сховішчамі інфармацыі, распрацаваны інтэрфейсы і рэалізацыі разборшчыкаў файлаў разнастайных фарматаў і канвертацыю іх у структуры канкрэтных біялагічных аб’ектаў. Зараз падтрымліваюцца фарматы асноўных сховішчаў біялагічнай іфармацыі: PDB, GenBank, EMBL, FASTA, SWISSPROT. Характэрна тое, што заданнем некалькіх параметраў даследчык можа фільтраваць даныя і працаваць толькі з неабходнай яму інфармацыяй. Для больш складаных і нетрывіяльных задач распрацоўчык вольны пашырыць існуючы ППІ. Прычым, пры ўмове якаснага і карыснага пашырэння, гэтыя змены могуць быць дададзеныя ў праект на аснове адкрытасці яго праграмных кодаў і потым паўторна выкарыстаны іншымі даследчыкамі. Таму бібліятэка вельмі дынамічна пашыраецца.
Яшчэ адной магчымасцю бібліятэкі з’яўляецца наяўнасць рэалізацый эфектыўных алгарытмаў параўнання паслядоўнасцяў і структур на аснове метадаў дынамічнага праграміравання і схаваных маркаўскіх мадэляў. Гэтая функцыянальнасць размешчаная ў пакетах org.biojava.bio.dp і org.biojava.bio.alignment. Найбольш вядомымі з прадстаўленых алгарытмаў з’яўляюцца алгарытмы Баўма-Вэлша, Нідлмана-Вунша, Сміта-Ўотэрмана. Безумоўна, гэта вельмі карысная асаблівасць, бо скарачае значны час на распрацоўку і тэсціраванне праграмы.
Наступнай важнай магчымасцю бібліятэкі BioJava з’яўляецца магчымасць візуалізацыі структур і праца з шэрагам знешніх візуалізатараў. Гэтая функцыянальнасць знаходзіцца ў пакеце org.biojava.bio.gui і ўкладзеных у яго. Адным са знешніх візуалізатараў, працу з якім забяспечвае BioJava і адным з найвядомейшых, з’яўляецца Java аплет Jmol.
Нарэшце, адной з найважнейшых магчымасцяў бібліятэкі з’яўляецца праца з базамі даных. Спецыяльна распрацаванай базай для працы з біялагічным аб’ектамі на аснове існуючых рэляцыйных баз даных, з’яўляецца BioSQL. Яна з’яўляецца агульнай мадэллю для біялагічных аб’ектаў і для працы з ёю існуюць ППІ на розных папулярных мовах, сярод якіх прадстаўлены Python, Java, Ruby, Perl. Шэраг баз таксама прадстаўлены шырокім спектрам: PostgreSQL, MySQL, Oracle, HSQLDB.
Трэба заўважыць, што хоць BioJava і з’яўляецца найбольш дарослай і вялікай бібліятэкай, аднак пад уплывам дынамічнага развіцця новых моў усё больш дынамічна развіваюцца і іншыя бібліятэкі, такія як BioRuby, BioPerl, BioPython. Гэта дазваляе пашырыць выкарыстанне такі ППІ сярод вялікай колькасці даследчыкаў, бо знікае неабходнасць засвойваць нейкую канкрэтную мову, а ёсць магчымасць знайсці адпаведнік на тое мове, навыкі працы з якой найбольш развітыя.
Глава 4 ПРАКТЫЧНАЕ ВЫКАРЫСТАННЕ БІБЛІЯТЭКІ BIOJAVA
§1 Пастаноўка задачы
Уваходныя даныя: файлы, у якіх змяшчаюцца апісанні паслядоўнасцяў ДНК чалавечага геному
Выхадныя даныя: пара найбольш блізкіх паслядоўнасцяў з гэтых файлаў.
Неабходна знайсці пару найбольш блізкіх паслядоўнасцяў з дапамогай бібліятэкі BioJava.
§2 Усталёўка бібліятэкі
Перад тым, як пачынаць працу з бібліятэкай і напісаннем праграмнага коду, відавочна, трэба зрабіць яе прымяняльняй. Бібліятэка распаўсюджваецца, як набор jar файлаў, таму ў пачатку неабходна скачаць архіў з праграмай і распакаваць файлы бібліятэкі з яго. Для распрацоўкі на платформе Java неабходны так называемы Java Development Kit (JDK) – мінімальны набор распрацоўшчыка. Таму неабходна праінсталіраваць і яго, пры ўмове таго, што ён не ўсталяваны. Для працы з апошняй версіяй BioJava 1.6.1 патрэбны JDK ад версіі 1.5 і вышэй. Для таго, каб выкарыстоўваць бібліятэку неабходна дадаць яе файлы ў гэтак званы CLASSPATH. Можна выстаўляць яго, як пераменную асяроддзя, а можна і перадаваць як параметр пры запуску віртульнай машыны Java (Java Virtual Machine). Ускарыстаемся першым варыянтам:
export CLASSPATH=/home/kotuk/biojava-live.jar:/home/kotuk/bytecode.jar:
/home/kotuk/commons-cli.jar:
/home/kotuk/commons-collections-2.1.jar:
/home/kotuk/commons-dbcp-1.1.jar:
/home/kotuk/commons-pool-1.1.jar:.
Як можна заўважыць, акрамя непасрэдна класаў бібліятэкі, мы павінны ўключыць і архівы бібліятэк, ад якіх залежыць BioJava. У асноўным гэта часткі вядомага праекта з адкрытым праграмным кодам Apache Jakarta Commons. З-за таго, што BioJava таксама з'яўляецца праектам з актрытым праграмным кодам, то мы маем магчымасць ускарыстацца кодамі класаў і сабраць праект самім. Але для канкрэтнай задачы гэта не з'яўляецца неабходным, таму праграмныя коды ў асноўным выкарыстоўваюцца для глыбейшага разумення механізму працы бібліятэкі.
З цягам часу з ростам памеру і складанасці праектаў на Java, эфектыўная распрацоўка стала немагчымай без гэтак званых інтэграваных асяроддзяў распрацоўкі (Integrated Development Environment, IDE). Яны дазваляюць зручна працаваць з кодам, збіраць і запускаць праекты без пераключэння паміж вокнамі, праглядаць дакументацыю, працаваць з сістэмамі кантролю версій. Аднымі з найбольш вядомых IDE для Java з'яўляюцца Eclipse, IntelliJ Idea, NetBeans. У гэтай працы выкарыстоўваецца асяроддзе IntelliJ Idea [9]. Гэты прадукт з'яўляецца платным, але прадстаўляецца бясплатны тэставы тэрмін даўжынёй у месяц.
Праца ў ёй пачынаецца са стварэння праекта. Пры яго стварэнні прадастаўляецца выбар са спісу гатовых шаблонаў Java-праграм, але з-за спецыфічнасці задач біяінфарматыкі, мы выбіраем пусты праект і пакідаем усе наладкі па змоўчанні. Ствараем галоўны клас нашай праграмы BioJavaProgram. У выніку атрымоўваем экран працоўнай вобласці на малюнку 4.1.
 Малюнак 4.1. Працоўнае асяроддзе IDE Idea
Малюнак 4.1. Працоўнае асяроддзе IDE Idea
§3 Выбар алгарытмаў
Агульнапрынятым падыходам для вырашэння задач папарнага выраўнівання нейкіх зададзеных паслядоўнасцяў з’яўляецца выкарыстанне дакладных алгарытмаў Нідлмана-Вунша і Сміта-Ўотэрмана. Адрозненне гэтых алгарымтаў заключаецца ў вобласці выраўнівання паслядоўнасця: Нідлман і Ванш вырашылі задачу глабальнага выраўнівання ў той час, як Сміт і Ўотэрман вырашылі задачу лакальных выраўніванняў. Менавіта алгарытм апошніх і пакладзены ў аснову такога вядомага праграмнага сродка, як BLAST. Ніжэй будзе паказана, як з дапамогай бібліятэкі ППІ BioJava можна з лёгкасцю вырашыць гэтую задачу. Неабходна заўважыць, то такая магчымасць з’явілася толькі ў версіі 1.5 бібліятэкі.
Ідэя дакладных алгарытмаў заключаецца ў выкарыстанні матрычнага прадстаўлення графа рэдагавання, гэта значыць такога графа, які пакрывае аперацыі ўстаўкі, выдалення, замены і адкрыцця разрыву. З дапамогай дынамічнага праграміравання вылічваюцца элементы матрыцы, якія ацэньваюць ацэнкі адпаведных аперацыі. Шлях з найбольшай ацэнкай(ці найменшым штрафам) дае нам найлепшае выраўніванне, якое можа быць атрымана праходам назад па элементах матрыцы. Яшчэ адным параметрам алгарытмаў з'яўляецца матрыца замен, якая прадугледжвае розныя штрафы на замену аднаго сімвала другім у паслядоўнасці на аснове статыстычных біялагічных даных.
Для выкарыстання гатовых рэалізацый дастаткова толькі задаць штрафы на аперацыі (устаўкі, разрыву, замены), матрыцу штрафаў замены сімвалаў і паслядоўнасці, якія мы жадаем параўнаць.
§4 Напісанне праграмы пошуку найбліжэйшых паслядоўнасцяў
Код праграмы разбіваецца на некалькі частак. У пачатку мы будуем аб'ект інтэрфейса SequenceAlignment, які будзе адпавядаць выбранаму намі алгарытму Нідлмана-Ванша. Для гэтага мы ініцыялізуем матрыцу замен (ствараем аб'ект класа sub>stitutionMatrix) на аснове файла матрыцы замен NUC-4.4 і выбранага алфавіту (у нашым выпадку гэта алфавіт ДНК). Гэтая і іншыя матрыцы даступны для скачвання ў Інтэрнэт па адрасе ftp://ftp.ncbi.nlm.nih.gov/blast/matrices. Акрамя гэтага задаем памеры штрафаў на супадзенне, замену, устаўку, выдаленен і працяг разрыву. Цяпер аб'ект>
sub>stitutionMatrix matr = new sub>stitutionMatrix(DNATools.getDNA(), new File(MATRIX_PATH));
SequenceAlignment aligner = new NeedlemanWunsch(0, 4, 2, 2, 2, matr);
Пасля гэтага дадаем функцыю getSequence, у якую параметрам перадаецца імя файла, з якога мы жадаем атрымаць паслядоўнасць. Нарэшце, дадаем функцыю getBestSeq, параметрам у якой выступае ініцыялізаваны намі раней выраўнівацель aligner. У ёй мы перабіраем усе магчымыя пары паслядоўнасцяй і выбіраем тую з іх, якая дае нам найлепшы вынік. Потым выводзім гэтае выраўніванне на экран.
Аналагічным чынам ініцыялізуем аб'ект-выраўнівацель і для алгарытму Сміта-Ўотэрмана. З-за таго, што абодва алгарытмы наследуюцца ад аднаго інтэрфейсу і маюць аднолькавую сігнатуру канструктараў змены неабходныя для стварэння новага аб'екту мінімальныя. Гэтаксама знаходзім пару з найлепшым выраўніваннем і вяртаем яе, а пасля ў галоўнай праграме выводзім гэтае выраўніванне на экран.
§5 Тэсціраванне праграмы пошуку найбліжэйшых паслядоўнасцяў
Для тэсціравання напісанай праграмы былі ўзяты файлы 16 паслядоўнасцяў ДНК чалавечага геному. Фарамат файлаў быў наступным: у першым радку змяшчалася назва канкрэтнага кавалка ў NCBI, у астатніх радках ішло непасрэднае апісанне паслядоўнасці ДНК у сімвалах алфавіту {A, C, G, T}, што адпавядае адэніну, цытазіну, гуаніну і тыяніну. Даўжыня паслядоўнасцяў была рознай і вагалася ад 220 да 720 сімвалаў. Усе гэтыя паслядоўнасці змяшчаліся ў каталог /home/skhamenk/dna, а ў праграме ўказваўся шлях да гэтага каталога ў глабальнай канстанце SEQUENCE_PATH. І ўжо адтуль праграма выбірала ўсе файлы і разбірала іх для далейшага выкарыстання.
У выніку выканання праграмы былі атрыманыя два найлепшых выраўніванні: адзін для алгартыму Нідлмана-Ванша, другі для алгарытму Сміта-Ўотэрмана. Як і можна было чакаць, выраўніванні не супалі і былі атрыманыя для розных паслядоўнасць. Гэта выклікана тым, што алгарытм Нідлмана-Ванша больш улічвае глабальнае падабенства паслядоўнасцяў, у той час як алгарытм Сміта-Ўотэрмана сканцэнтраваны на пошуку лакальных выраўніванняў.
У выніку працы першага алгарытму наступнае выраўніванне паслядоўнасцяў было выяўлена, як найлепшае: паслядоўнасці з кодамі BG560803 D104 і BG560804 D106 найбольш адпавядаюць адна адной пры глабальным параўнанні. Атрыманая ацэнка -68.0 з'яўляецца невысокай, але гэта выклікана тым, што паслядоўнасці для тэсціравання выбіраліся выпадковым чынам. Час выканання быў меншым за секунду.
У выніку працы другога алгарытму было атрымана, што паслядоўнасці з кодамі BG560814 D221 і BG560810 D215 найлепш адпавядаюць адна адной у лакальным плане. Іх даўжыні адпаведна складаюць 699 і 398 сімвалаў. Атрыманая ацэнка велічынёй у 764 сведчыць пра наяўнасць падобных кавалкаў у паслядоўнасцях.
Вывад выраўніванняў ажыццяўляўся з дапамогай рэалізавана ў BioJava метада getAlignmentString(). Ён змяшчае ўсю неабходную інфармацыю пра выраўніванне і праводзіць простую візуалізацю зробленых дзеянняў па рэдагаванні паслядоўнасцяў у аптымальным рашэнні. Вывад ажыццяўляецца ў тэкставым фармаце і прадстаўлены на малюнку 4.2. З-за выкарыстання тэхнікі дынамічнага праграміравання складанасць алгарытмаў складае O(m * n), дзе m i n – даўжыні паслядоўнасцяў для выраўнівання. Гэта дазваляе нам рашаць задачу за вельмі кароткі час на сучаснай тэхніцы.
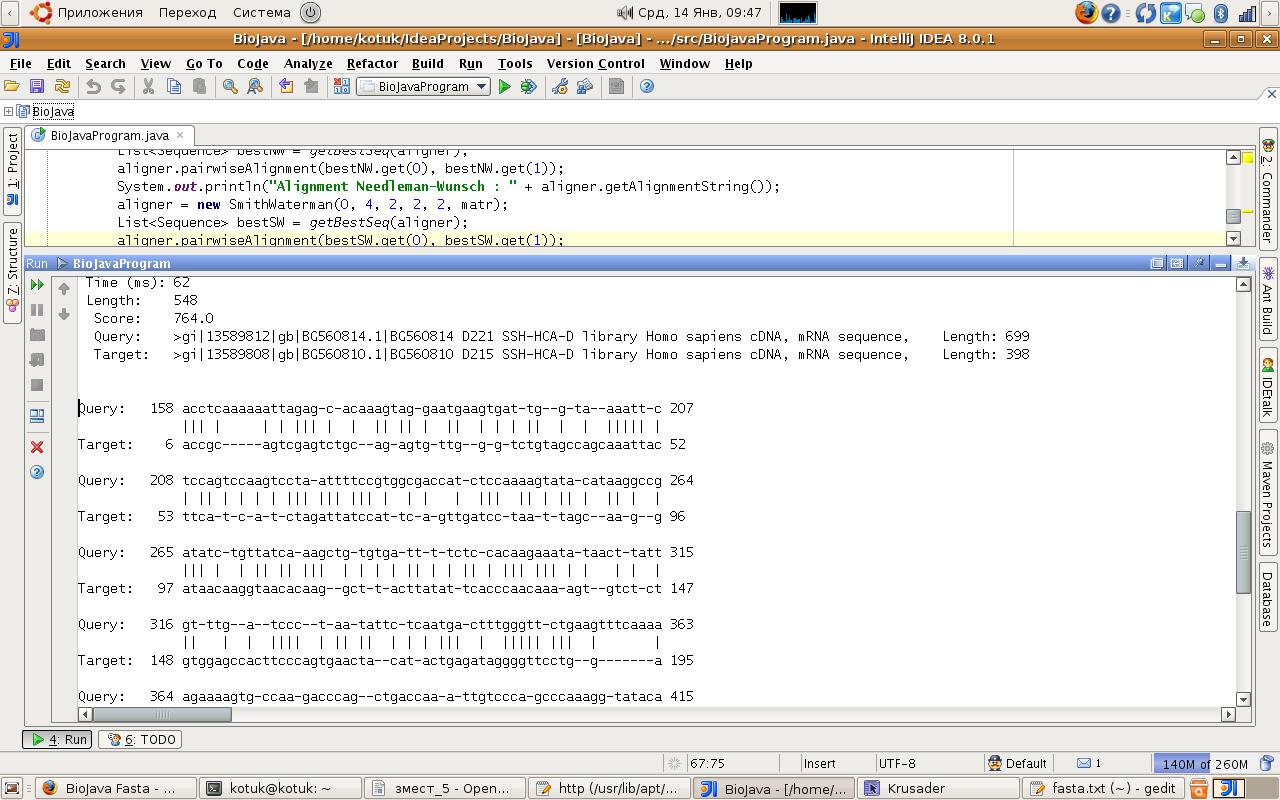
Малюнак 4.2 Вывад выраўнівання найбольш падобных паслядоўнасцяў
Такім чынам, былі знойдзены найбліжэйшыя адпаведнікі сярод уваходных паслядоўнасцяў і зроблена простая візуалізацыя іх падабенства. Гэта было зроблена пры выкарыстанні магчымасцяў мовы праграміравання Java і бібліятэкі для распрацоўкі праграм для біяінфарматыкі BioJava. Лістынг праграмы прыведзены ў Дадатку 1.
ЗАКЛЮЧЭННЕ
Такім чынам, у дадзенай працы была праведзена класіфікацыя праграмных сродкаў і паказаны асноўныя сферы прымянення інфармацыйных тэхналогій у біяінфарматыцы. Відавочна, што гэтая галіна зараз атрымоўвае ўсё большую і большую ўвагу. Не гледзячы на тое, што біяінфарматыка адносна маладая галіна інфарматыкі і малекулярнай біялогіі, але колькасць выданняў, навуковых лабараторый, гатовых праграмных сродкаў і праграмных бібліятэк сведчаць пра важнасць тых задач, якія стаяць перад ёю.
У першай главе дадзенай працы былі разгледжаны асноўныя прымяненні інфармацыйных тэхналогій у зборы, захоўванні і апрацоўцы інфармацыі. Былі апісаны асноўныя віды і тыпы сховішчаў біялагічнай інфармацыі ў залежнасці да самога тыпу інфармацыі. Была ілюстратыўна паказана і характарызавана праца з такімі праграмнымі сродкамі, як візуалізатары біялагічнай інфармацыі. Сцісла ахарактарызаваны такі клас праграм, як пашуковыя сістэмы, існаванне якога цесна звязанае з галоўным элементам біяінфарматыкі – сховішчамі біялагічнай інфармацыі.
У другой главе былі апісаны інфарматычныя падыходы да задач супастаўлення з узорам і прымянімасць іх да рашэння канкрэтных задач біяінфарматыкі. Былі апісаны галоўныя праграмныя сродкі, якія выкарыстоўваюцца зараз даследчыкамі для параўнання лінейных прасторавых структур бялкоў.
Трэцяя глава была прысвечана апісанню адной з найвядомейшых і карысных бібліятэк для стварэння праграм па рашэнні задач біяінфарматыкі BioJava. Былі падрабязна апісаны і патлумачаны асноўныя магчымасці гэтай бібліятэкі. А таксама падкрэслена роля той свабоды, якую даюць такія праграмныя сродкі распрацоўшчыку для рэалізацыі яго ідэй.
У чацвёртай апошняй главе з дапамогай апісанай раней бібліятэкі BioJava была выканана практычная задача вынаходжання найбольш падобных паслядоўнасцяў з нейкага мноства. У гэтай главе былі падрабязны апісаны крокі па ўсталёўцы, наладцы і выкарыстанні гэтай бібліятэкі.
Такім чынам, відавочная немагчымасць існавання біяінфарматыкі без наяўнасці інфармацыйных тэхналогій. І пры існуючых каласальных тэмпах развіцця інфарматыкі і камп'ютэрнай тэхнікі можна казаць пра такое ж хуткае развіццё і біяінфарматыкі, што прывядзе ў выніку да эфектыўнага вырашэння практычных задач біялогіі і медыцыны.
СПІС ВЫКАРЫСТАНАЙ ЛІТАРАТУРЫ
[1] Protein Data Bank [Электронны рэсурс] . - Рэжым доступу: http://www.rcsb.org . - Дата доступу: 14.01.2009
[2] Bergeron B. Bioinformatics computing. 1st ed. Upper Saddle River, NJ: Prentice Hall PTR, 2002, 395p.
[3] Візуалізатар Aceleryss [Электронны рэсурс] . - Рэжым доступу: http://www.rcsb.org . - Дата доступу: 14.01.2009
[4] NCBI Map Viewer [Электронны рэсурс] . - Рэжым доступу: http://www.ncbi.nlm.nih.gov/projects/mapview/ . - Дата доступу: 14.01.2009
[5]NCBI Entrez [Электронны рэсурс] . - Рэжым доступу: http://www.ncbi.nlm.nih.gov/Entrez/ . - Дата доступу: 14.01.2009
[6]BLASTN [Электронны рэсурс] . - Рэжым доступу: http://www.ncbi.nlm.nih.gov/Entrez/ . - Дата доступу: 14.01.2009 [7]3DSS[Электронны рэсурс] . - Рэжым доступу: http://cluster.physics.iisc.ernet.in/3dss/ . - Дата доступу: 14.01.2009
[8]BioJava[Электронны рэсурс] . - Рэжым доступу: http://biojava.org/ . - Дата доступу: 14.01.2009
[9]IntelliJ Idea[Электронны рэсурс] . - Рэжым доступу: http://www.jetbrains.com/idea/ . - Дата доступу: 14.01.2009
ДАДАТАК 1
Лістынг праграмы BioJavaProgram
import org.biojava.bio.symbol.IllegalSymbolException;
import org.biojava.bio.seq.DNATools;
import org.biojava.bio.seq.Sequence;
import org.biojava.bio.alignment.sub>stitutionMatrix;
import org.biojava.bio.alignment.SequenceAlignment;
import org.biojava.bio.alignment.NeedlemanWunsch;
import org.biojava.bio.alignment.SmithWaterman;
import java.io.*;
import java.util.List;
import java.util.ArrayList;
/**
* @author Siarhei Khamenka
*/
public class BioJavaProgram {
public static final String MATRIX_PATH = "/home/kotuk/NUC44.MAT";
public static final String SEQUENCES_PATH = "/home/kotuk/fasta/";
static Sequence getSequence(String fileName) {
try {
BufferedReader br = new BufferedReader(new FileReader(SEQUENCES_PATH + fileName));
String name = br.readLine(), st, seq = "";
while ((st = br.readLine()) != null) {
seq += st;
}
br.close();
return DNATools.createDNASequence(seq, name);
} catch (IOException e) {
e.printStackTrace();
} catch (IllegalSymbolException e) {
e.printStackTrace();
}
return null;
}
static List<Sequence> getBestSeq(SequenceAlignment>
String[] seqFiles = new File(SEQUENCES_PATH).list();
Sequence bestQuery = null, bestTarget = null;
double maxRes = Double.MIN_VALUE;
for (int i = 0; i < seqFiles.length; i++) {
Sequence query = getSequence(seqFiles[i]);
for (int j = i + 1; j < seqFiles.length; j++) {
Sequence target = getSequence(seqFiles[j]);
double res =>
if (res > maxRes) {
bestQuery = query;
bestTarget = target;
}
}
}
List<Sequence> res = new ArrayList();
res.add(bestQuery);
res.add(bestTarget);
return res;
}
public static void main(String[] args) throws Exception {
sub>stitutionMatrix matr = new sub>stitutionMatrix(DNATools.getDNA(), new File(MATRIX_PATH));
SequenceAlignment aligner = new NeedlemanWunsch(0, 4, 2, 2, 2, matr);
List<Sequence> bestNW = getBestSeq(aligner);
>
System.out.println("Alignment Needleman-Wunsch : " +>
>
List<Sequence> bestSW = getBestSeq(aligner);
>
System.out.println("Alignment Smith-Woterman : " +>
}
}
1