Исследование соотношения в мышцах С- и Х-белков в норме и при патологии
Федеральное агентство по образованию
Пензенский государственный педагогический университет
им. В.Г. Белинского
Факультет Естественно-географический
Кафедра Биохимии
Дипломная работа
Исследование соотношения в мышцах С- и Х-белков в норме и при патологии
Студент _________________________________________ Швецов Ю.А.
подпись
Научный руководитель
Зав. лаб. ИТЭБ РАН, д.б.н., профессор______________Подлубная З.А.
подпись
Научный руководитель
доц. каф. биохимии, к.б.н.__________________________ Соловьев В.Б.
подпись
К защите допустить. Протокол № от «____» ___________200_г.
Зав. кафедрой _____________________________________ Генгин М.Т.
подпись
Пенза, 2008 г.
содержание
Список использованных сокращений……………………...………………….…4
ВВЕДЕНИЕ………………………………………………………………...……..5
I. обзор литературы…………………………………………………...…………..8
Глава 1. САРКОМЕРНЫЕ ЦИТОСКЕЛЕТНЫЕ БЕЛКИ СЕМЕЙСТВА тайтинА…………………………………………………………………………….8
1.1 Структура молекулы тайтина и его функции…………….………………8
1.2 Структура и функции молекул С-белка, Х-белка и Н-белка……..…...10
1.3 Белки семейства тайтина в норме, при адаптации и патологии…….….14
Глава 2. Амилоидозы………………………………………..…….………….…16
2.1. Актуальность проблемы……………………………………………..…...16
2.2. История изучения амилоидозов….……………………………………....17
2.3. Современные представления о строении и формировании амилоидных фибрилл…………………………………………………………………………..19
2.4. Изучение амилоидных фибрилл in vitro……………………………...….25
Патологические проявления амилоидозов……………………………...30
ЦЕЛЬ И ЗАДАЧИ ИССЛЕДОВАНИЯ………………………………………....32
II. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ………………………………………....32
Глава 3. МАТЕРИАЛЫ И МЕТОДЫ…………………………………..……....33
3.1. Экспериментальный и клинический материал…………………...……..33
3.2. Выделение и очистка белковых препаратов…………………...………..33
3.2.1. Очистка С-белка, Х-белка и Н-белка…………...………..........………....35
3.2.2. Выделение тайтина из скелетных мышц………………………...……....35
3.3. Определение концентрации белковых препаратов…………………......34
3.4. Проверка чистоты белковых препаратов………………………...……...35
3.5. Условия формирования амилоидных фибрилл…………………...…….36
3.6. Микроскопические исследования……………………………...………...37
3.6.1 Электронная микроскопия……………………………………………......37
3.6.2. Поляризационная и флуоресцентная микроскопия………………..…...37
3.7. Спектральные методы……………………………………………...……..37
3.7.1. Метод кругового дихроизма…………………………………...………....37
3.7.2. Метод флуоресцентного анализа…………………………………...…....38
3.7.3. Спектрофотометрический метод……………………………..………….38
III. Результаты и обсуждение………………………..………………….……...40
Глава 4. образование амилоидных фибрилл белками семейства тайтина….40
Электронно-микроскопическое изучение агрегационных свойств молекул тайтина, Х-белка, С-белка и Н-белка скелетных мышц кролика…………………………………………………………..……………….40
4.2. Электронно-микроскопическое изучение агрегационных свойств Аβ(25-35)-пептида в сравнении с агрегацией молекул Х-белка…………….47
Подтверждение амилоидной природы агрегатов, образуемых белками семейства тайтина (тайтина, Х-белка, С-белка и Н-белка) при их взаимодействии со специфическими красителями на амилоиды Конго красным и тиофлавином Т…………………………………………………......49
Изучение вторичной структуры тайтина и белков его семейства до и после образования амилоидных фибрилл ……………………....................….53
4.5. Скорость образования амилоидных фибрилл Х-белка..………...…..….55
Образование амилоидных фибрилл С-белком миокарда человека при ДКМП и С-белком миокарда кролика………………………..……………...…57
Список литературы………………………………………………………….....61
Список использованных сокращений
ДКМП дилатационная кардиомиопатия
ДСН додецилсульфат натрия
ДТТ дитиотреитол
КД круговой дихроизм
кДа килодальтон
КК Конго красный
ЛММ легкий меромиозин
МДа мегадальтон
мкм микрометр
нм нанометр
ТТ тиофлавин Т
УФ ультрафиолет
ЭГТА этиленгликоль бис- (β-аминоэтиловый эфир N,N,N',N-тетрауксусной кислоты)
ЭДТА этилендиаминтетраацетат
FnIII фибронектин-3-подобный домен
IgC2 иммуноглобулин-С2-подобный домен
PMSF фенилметилсульфонилфторид
λ длина волны
μ ионная сила
введение
Амилоидозы – большая группа конформационных заболеваний, которая характеризуется отложениями белка в виде нерастворимых фибрилл в разных органах и тканях, образующихся в результате наследственного или приобретенного нарушения сворачивания белков (Tan & Perys, 1994; Uversky & Fink, 2004). Их накопление разрушает структуру и функционирование органов и тканей, приводя к болезни и летальному исходу. Амилоидные отложения найдены при болезни Альцгеймера, Паркинсона, Дауна, диабете второго типа, наследственной амилоидной полинейропатии, системном амилоидозе и др. (Tan & Perys, 1994; Goedert, 2001; Dobson, 2001).
Известно много белков, образующих амилоидные фибриллы, таких как тау-белок, Аβ-пептид, ацилфосфатаза, миоглобин, амилин, транстиретин и другие (Guijarro et al., 1998; Chiti et al., 1999; Fandrich et al., 2001; Uversky & Fink, 2004). Несмотря на различие в белках-предшественниках амилоидов, образуемые ими амилоидные фибриллы имеют общие свойства: β-складчатую структуру с отдельными β-слоями, ориентированными параллельно главной оси фибриллы; нерастворимость in vivo; специфическое связывание с красителями Конго красным и тиофлавином Т (Klunk et al., 1989; Krebs et al., 2005). Важно отметить, что белки-предшественники амилоидов претерпевают трансформацию типа "α-спираль – β-складчатость", необходимую для образования амилоидных фибрилл (Uversky & Fink, 2004).
К сожалению, процессы, лежащие в основе аномальной агрегации белка и ее патологического проявления при болезнях, изучены еще недостаточно. Выяснение молекулярных механизмов амилоидозов, установление белковой природы депозитов и их свойств, развитие терапевтических методов лечения и предупреждения этих заболеваний, а также разработка их прижизненной диагностики являются актуальными задачами. Успешное решение этих задач во многом зависит от фундаментальных знаний амилоидогенеза: выяснения свойств амилоидов разных белков, знания факторов, регулирующих их образование и разрушение, их эффектов на жизнедеятельность разных клеток и т.д. В наибольшей мере это относится к мышечным амилоидозам как к наименее изученным. Амилоидные отложения найдены при кардиомиопатиях, миокардитах и миозитах в мышцах и кровеносных сосудах, однако их белковая природа до сих пор неизвестна.
Данная работа посвящена изучению способности белков семейства тайтина формировать амилоидные фибриллы. В настоящее время известна большая группа белков семейства тайтина (тайтин, С-белок, Х-белок, Н-белок и другие), относящихся к саркомерным белкам поперечно-полосатых мышц позвоночных. Они связаны с миозин-содержащими (толстыми) нитями и составляют 15% от общего количества белка в саркомере. С-белок в быстрых волокнах скелетных мышц и его изоформа Х-белок в медленных волокнах (Yamamoto & Moos, 1983; Starr & Offer, 1983) располагаются на поверхности толстых нитей с периодом 43 нм (Bennett et al., 1986; Soteriou et al1., 1993), связываясь одновременно с тайтином и миозином. Тайтин является продольным элементом саркомерного цитоскелета поперечно-полосатых мышц позвоночных.
Исследования показали, что тайтин и белки его семейства могут выполнять разные функции в саркомере. Предполагается, что они играют существенную роль в миогенезе при сборке толстых нитей и формировании структуры саркомера, участвуют в регуляции актин-миозинового взаимодействия при мышечном сокращении и в процессах сигнализации. Наличие 90% β-складчатой структуры в этих белках создает возможность формирования ими амилоидов. Изучение способности белков семейства тайтина формировать амилоидные фибриллы представляет большой интерес в связи с их возможным участием в развитии амилоидозов. Ранее при изучении агрегационных свойств белков, связанных в саркомере с миозиновыми нитями, было обнаружено, что один из них (Х-белок) образует in vitro спирально скрученные ленточные фибриллы (Bennett et al., 1985). Авторы указали на визуальное сходство этих структур с амилоидными фибриллами, образуемыми Аβ-пептидом в мозге при болезни Альцгеймера. Однако разные структурные параметры фибрилл Х-белка заставили авторов сомневаться в этом предположении и, возможно, поэтому тестирование их на амилоидогенность не было проведено. Эти наблюдения и высокое содержание β-складчатой структуры стимулировали наше исследование амилоидной природы агрегатов, образуемых Х-белком и другими саркомерными белками семейства тайтина.
I. ОБЗОР ЛИТЕРАТУРЫ
Глава 1. САРКОМЕРНЫЕ ЦИТОСКЕЛЕТНЫЕ БЕЛКИ СЕМЕЙСТВА ТАЙТИНА
Сократительным аппаратом мышечной клетки является миофибрилла, которая состоит из наименьших сократительных единиц – саркомеров. Саркомеры расположены друг за другом вдоль оси миофибриллы. Каждый саркомер содержит упорядоченную систему цитоскелетных и сократительных белков и имеет длину 2.5–3.0 мкм (Squire, 1981; Шубникова и др. 2001). Основными сократительными белками саркомера являются миозин, актин и тайтин. Молекулы актина образуют тонкие (актиновые) нити, в состав которого входят также тропомиозин и тропонин. Толстые нити скелетных мышц кроме миозина содержат также другие белки, такие как тайтин, С-белок, Х-белок, Н-белок и др. (рис. 1).
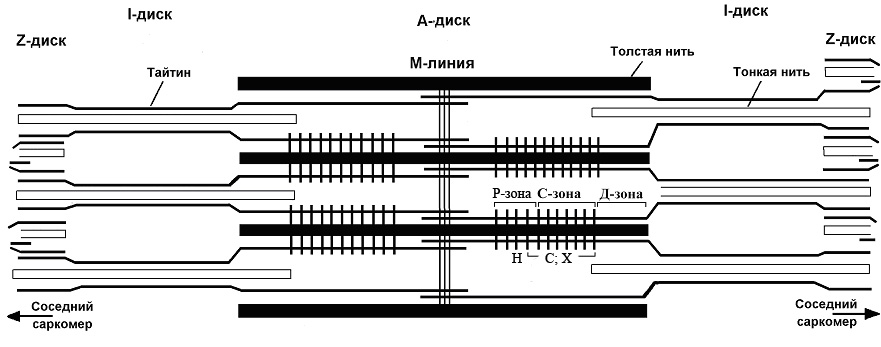
Рис.1. Модифицированная схема саркомера (Gregorio et al., 1999).
1.1 Структура молекулы тайтина и его функции
Тайтин (= коннектин, титин) – гигантский эластичный белок скелетных и сердечных мышц млекопитающих (Maruyama et al., 1977; Wang et al., 1979) с молекулярным весом более 3 МДа (Labeit & Kolmerer, 1995). Он был открыт независимо двумя группами исследователей методом ДСН-гель-электрофореза (Wang et al., 1979; Maruyama et al., 1981). Тайтин является третьим по количеству (после актина и миозина) компонентом саркомера поперечно-полосатых мышц позвоночных. Молекулы тайтина длиной около 1 мкм и шириной 3–4 нм (Trinick et al., 1984; Itoh et al., 1986; Nave et al., 1989; Soteriou et al., 1993; Suzuki et al., 1994; Houmeida et al., 1995; Tskhovrebova & Trinick, 1997) перекрывают половину саркомера от М-линии до Z-линии (рис. 1), простираясь через I- и А-диски саркомера, формируя третью филаментную систему в миофибриллах (Fürst et al., 1988). NH2-концы молекулы тайтина из смежных саркомеров перекрываются в Z-линии, а СООН-концы перекрываются в М-линии. На каждую половину саркомера приходится по шесть молекул тайтина, которые объединены в А-диске саркомера (Trinick 1981; Liversage et al., 2001) и, предположительно, разделяются на пары или на индивидуальные молекулы в I-диске саркомера (Funatsu et al., 1993).
Исследования последних лет показали, что этот гигантский полипептид имеет различное строение в разных зонах саркомера, что вносит свой вклад в их архитектуру и функционирование. Бóльшая часть (~90%) молекулы тайтина состоит из повторяющихся иммуноглобулин-С2-подобных (IgС2) и фибронектин-3-подобных (FnIII) доменов (Labeit & Kolmerer, 1995) с β-складчатой структурой, содержащих около ста аминокислотных остатков (Improta et al., 1996; Muhle-Goll et al., 1998) (рис. 2). Кроме этих доменов тайтин содержит уникальные последовательности, расположенные по всей длине его молекулы: киназный домен вблизи М-линии саркомера; два участка связывания с кальпаиновой протеазой (р94) в I-диске и М-линии саркомера, а также эластичные N2B, N2A и PEVK элементы в I-диске саркомера. В концевых областях молекулы тайтина расположены фосфорилируемые участки.
1.2 Структура и функции молекул С-белка, Х-белка и Н-белка
Предположение о присутствии белков немиозиновой природы в структуре толстых, или миозин-содержащих нитей на основании электронно-микроскопических и рентгеновских исследований высказывались давно (Draper & Hodge 1949; Franzini-Armstrong & Porter 1964; Pepe 1967; Hanson et al., 1971), однако до начала 70-х годов природа этих белков была неизвестна. С-белок, Х-белок и Н-белок были обнаружены с помощью ДСН-гель-электрофореза как минорные примеси в препаратах миозина скелетных мышц кролика (Starr & Offer, 1971). В настоящее время эти белки охарактеризованы, и их количество в саркомере составляет около 5% от массы миозина.
С-белок впервые был выделен из скелетных мышц кролика (Offer et al., 1973). Его количество составляет около 2% от массы миофибрилл и около 4% от массы толстой нити.
Показана способность сердечного С-белка фосфорилироваться разными протеинкиназами в условиях in vivo и in vitro (Jeacocre & England, 1980; Hartzell & Titus, 1982; Hartzell & Glass, 1984; Hartzell, 1985; Lim et al., 1985; Gautel et al., 1995; Mohamed et al., 1998). Молекулярный вес С-белка скелетных мышц по данным ДСН-гель-электрофореза 135 кДа (Yamamoto & Moos, 1983; Starr & Offer, 1983), а по данным анализа нуклеотидной последовательности генов, кодирующих этот белок, составляет 128.1 кДа (Weber et al., 1993). Аминокислотный состав белка характеризуется довольно высоким содержанием пролина, что коррелирует с низким содержанием α-спирали в его молекуле. Методом седиментации показано, что С-белок связывается с высоким сродством с миозином (Callaway & Bechtel, 1981; Yamamoto & Moos, 1983). Электронно-микроскопические исследования подтвердили это связывание для С-белка: он декорирует реконструируемые миозиновые нити и паракристаллы легкого меромиозина (ЛММ) in vitro (Moos et al., 1975; Starr & Offer, 1978; Safer & Pepe, 1980; Фрейдина и др., 1980; Подлубная, 1981; Podlubnaya et al., 1990). Показана способность С-белка связываться с актином (Moos et al., 1976; Moos et al., 1978; Moos, 1981; Фрейдина и др., 1980) и тайтином (Koretz et al., 1993; Soteriou et al., 1993; Fürst et al., 1992; Freiburg & Gautel, 1996). При снижении ионной силы ниже физиологических значений (μ ≤ 0.1).
Х-белок содержится в грубых препаратах С-белка, выделенного из миозина хроматографией на ДЕАЕ-сефадексе (Starr & Offer 1982). Был разработан метод разделения Х- и С-белков с помощью колоночной хроматографии на гидроксиапатите (Starr & Offer 1982; Starr et al., 1979). Молекула Х-белка представляет собой гибкую ниточку длиной около 30–35 нм и диаметром 3–4 нм (Bennett et al., 1985). Молекулярный вес Х-белка скелетных мышц кролика по данным ДСН-гель-электрофореза составляют и 145–152 кДа (Yamamoto & Moos 1983; Starr & Offer 1983), а по данным анализа нуклеотидной последовательности генов, кодирующего этот белок, составляют 126.5 кДа (Weber et al., 1993).
Аминокислотный состав характеризуется высоким содержанием пролина, что соответствует низкому (4%) содержанию α-спирали.
Н-белок выделен из скелетных мышц кролика, точнее – из грубых препаратов С-белка, хроматографией на гидроксиапатите (Starr & Offer, 1982; 1983). Он составляет 0.3–0.4% от миофибриллярной массы. Молекула Н-белка содержит одну полипептидную цепь с молекулярным весом по данным ДСН-гель-электрофореза ~ 69–74 кДа (Starr & Offer, 1983), а по данным другого метода ~ 86 кДа (Vaughan et al., 1993). Молекулярный вес Н-белка скелетных мышц цыпленка и человека по данным анализа нуклеотидной последовательности генов, кодирующего этот белок, составляет 58.4 кДа и 51.9 кДа, соответственно (Vaughan et al., 1993). Аминокислотный состав Н-белка сходен с аминокислотным составом С-белка, но отличается от последнего более высоким содержанием пролина и аланина и более низким содержанием аспарагиновой кислоты, валина и лизина (Yamamoto, 1984). Молекула Н-белка содержит около 4% α-спирали (Starr & Offer, 1983). Н-белок взаимодействует с миозином и с актином (Starr & Offer, 1983).
Белки семейства тайтина могут участвовать в сборке толстых нитей и стабилизации их структуры. Известно, что С-белок, также как и тайтин (Wang & Wright, 1988), появляется в мышечной клетке на ранних стадиях миофибриллогенеза (Bahler et al., 1985). Имеются также данные о влиянии тайтина, С-белка и Х-белка на формирование миозиновых нитей in vitro (Koretz, 1979; Miyahara & Noda, 1980; Koretz et al., 1982; Вихлянцев 2005). Вероятно, что С-белок (вместе с тайтином) участвует в сборке и стабилизации структуры толстых нитей. Исследования in vitro показали, что Н-белок влияет на связывание С-белка с миозином (Yamamoto, 1984). Показано связывание С-белка и тайтина с актином в условиях in vitro. Предполагается, что физиологическое значение этого связывания в саркомерах скелетных мышц заключается в регуляции актин-миозинового взаимодействия. В экспериментах in vitro показано, что тайтин и С-белок активируют АТФазу актомиозина. Это влияние может быть обусловлено как модифицирующим действием С-белка и тайтина на миозиновый мостик, так и прямым их взаимодействием с актином (Moos et al., 1978, 1980; Moos, 1981; Muhle-Goll et all., 2001; Moolman-Smook et al., 2002; Squire et al., 2003). Х-белок и Н-белок также могут быть вовлечены в процесс мышечного сокращения, ингибируя АТФ-азную активность актомиозина (Yamamoto, 1984, Вихлянцев, 2005). В поддержку предположения о регуляторной роли С-белка в сократительной активности являются данные о фосфорилировании С-белка сердечной мышцы. В ранних исследованиях было показано, что уровень фосфорилирования С-белка в интактном сердце коррелирует со скоростью релаксации мышцы (Hartzell & Glass, 1984). In vitro фосфорилирование сердечного С-белка приводило к снижению его активирующего эффекта на АТФазу АМ (Hartzell, 1985). На основе полученных данных автором было сделано предположение, что С-белок участвует в механизме расслабления сердечной мышцы. Однако результаты дальнейших исследований показали, что фосфорилирование С-белка модулирует процесс сокращения сердечной мышцы, увеличивая доступность миозиновых головок к актину (Weisberg & Winegrad, 1996, 1998; Kunst et al., 2000; McClellan et al., 2001; Kulikovskaya et al., 2003; McClellan et al., 2004; Flashman et al., 2004). Действительно, согласно последним данным фосфорилирование сердечного С-белка приводит к усилению сократительной активности мышцы (Stelzer et al., 2006).
1.3. Белки семейства тайтина в норме, при адаптации и патологии
Структурно-функциональные свойства белков семейства тайтина в норме описаны выше. Эти белки являются важными компонентами сократительного аппарата мышечной клетки, участвуя в построении и стабилизации структуры саркомера. Влияние белков семейства тайтина на АТФазу актомиозина указывает на их важную регуляторную функцию в норме.
Поведение белков семейства тайтина при адаптации рассматривается на примере гибернации (зимней спячки). Показано, что у гибернирующего животного уменьшается площадь поперечного сечения мышечных волокон. Изменения структурно-функциональных свойств миозин-содержащих нитей скелетных мышц вносят вклад в подавление двигательной активности животного при зимней спячке (Лукоянова и др., 1996). В скелетных мышцах гибернирующих животных обнаружено снижение активирующего влияния тайтина и С-белка и увеличение ингибирующего влияния Х-белка на ферментативные и регуляторные свойства миозина (Вихлянцев и др., 2000; 2002).
Поскольку причину развития ДКМП большинство исследователей видит в повреждении сократительных структур миокарда, а условия микрогравитации приводят к "гипогравитационному мышечному синдрому" (Гуровский и др., 1975; Nemirovskaya et al., 2002), становится вполне понятным пристальное внимание исследователей к этим патологическим состояниям. Показано, что пребывание в условиях моделируемой микрогравитации приводит к уменьшению количества тайтина и Х-белка в m. soleus крыс и человека, что, наряду с другими изменениями в мышечном аппарате, будет вносить вклад в развитие "гипогравитационного мышечного синдрома" (Вихлянцев и др., 2006). Увеличение содержания тайтина в миокарде левого желудочка человека при ДКМП приводит к снижению уровня пассивного напряжения одиночных миофибрилл и волокон, что отражается на сократительной функции сердца (Макаренко и др., 2002, Макаренко, 2004).
Данные, полученные при исследовании свойств полифункциональных белков семейства тайтина в норме, при адаптации и заболеваниях дают основание предполагать, что изменения их структурно-функциональных характеристик может вносить вклад в развитие патологических процессов в мышцах. Выяснение роли тайтина, Х-белка, С-белка и Н-белка в патогенезе разных болезней является чрезвычайно актуальной задачей. В данной работе объектом нашего внимания являлись амилоидозы и, в частности, амилоидные свойства этих белков in vitro.
Глава 2. Амилоидозы
2.1. Актуальность проблемы
Амилоидозы – болезни, которые характеризуются отложениями нерастворимых фибрилл белка (амилоидных фибрилл) в разных органах и тканях, образующихся в результате наследственного или приобретенного нарушения сворачивания белков. Амилоидные отложения играют центральную роль в патогенезе болезней, от которых страдают миллионы пациентов (болезнь Альцгеймера, Паркинсона, Дауна, диабет II типа, наследственная амилоидная полинейропатия, системные амилоидозы, прионные амилоидозы и др.) (Uversky & Fink, 2004). Однако, процессы, лежащие в основе аномальной агрегации белка и ее патологического проявления при болезнях, изучены еще недостаточно.
Амилоидоз широко распространен среди многих представителей животного мира. Описаны первичные (идиопатические), вторичные (приобретенные), наследственные и старческие его формы (Виноградова, 1980). Амилоидные отложения могут достигать килограммов (как, например, фибриллярные скопления лизоцима в печени). Они найдены также в сердечной мышце при кардиомиопатиях, миокардитах и в скелетных мышцах при миозитах (Барсуков и др., 2005). При миокардитах (воспалительное поражение сердечной мышцы) амилоид имеет вид россыпи. Возможно тотальное поражение сердца или только предсердий, только желудочков или клапанов. При кардиопатическом амилоидозе амилоид откладывается в эндо-, мио- и эпикарде. Отложения амилоида в сердце приводят к резкому увеличению его размеров (амилоидная кардиомегалия). Оно становиться очень плотным, миокард приобретает сальный вид ("резиновый миокард"). Миозит с включенными тельцами (амилоидами) сопровождается изнурительными мышечными болями. Амилоидные отложения обнаружены в скелетных мышцах, в миокарде, и по ходу межмышечной соединительной ткани, а также в стенках сосудов и нервах. Мышцы становятся плотными, полупрозрачными (Виноградова, 1980).
Уже сейчас амилоидозы – главная причина смерти после сердечно-сосудистых и раковых заболеваний. Диагностика большинства из них посмертная. Генезис этого заболевания, при котором возможно поражение любых органов и тканей и, следовательно, возникновение разнообразной клинической симптоматики, остается до конца не изученным. Возможно, причиной является спонтанное развитие амилоидоза, или наследственная передача болезни. Патогенез амилоидоза не уточнен, клинические проявления весьма пестрые и не всегда четко очерчены, лечение малоэффективно и редко диагностируется при жизни. Выяснение молекулярных механизмов амилоидозов, установление белковой природы депозитов и их свойств, развитие терапевтических методов лечения и предупреждения этих заболеваний, а также разработка их прижизненной диагностики являются актуальными задачами.
2.2. История изучения амилоидозов
Первое описание амилоидоза у человека относится к XVII веку, когда Боне сообщил результаты наблюдения больного с абсцессом печени и громадной селезенкой, содержащей множество белых камней (саговая селезенка). Дальнейшую историю изучения амилоидоза можно разделить на три этапа. Начало первого этапа связано с именем венского патолога Рокитанского (1842 г.), открывшего "сальную болезнь", развивающуюся у больных туберкулезом, сифилисом, риккетсиозами. Позже, как выяснил Меккель (1853 г.), "сальная", или "холестериновая" болезнь обычно является второй болезнью и может поражать многие органы. В 1854 г. немецкий физиолог Вирхов на основании характерного прокрашивания патологических структур мозга йодом, решил, что образующиеся массы имеют углеводную природу – "подобны крахмалу" и ввел термин "амилоид", происходящий от латинского "амилум" и от греческого "амилон". Через несколько лет Фридрайх и Кекуле на основании химического анализа доказали белковую природу амилоидного вещества, однако термин "амилоид", "амилоидоз" сохранился до настоящего времени. Второй этап изучения амилоидоза относится к 20-м годам XX столетия, когда Бенхольд (1922 г.) предложил окраску амилоида Конго красным, обнаружив эффект двойного лучепреломления в поляризованном свете. Этот эффект указывает на тот факт, что амилоидные образования представляют собой упорядоченные микроскопические структуры. Данный метод впоследствии стал первым диагностическим тестом для определения амилоидов в клинической практике. В 1959 г. Коген и Калкинс с помощью электронной микроскопии установили, что все типы амилоида человека и экспериментальных животных имеют фибриллярную структуру (Sipe & Cohen, 2000). Амилоид оказался образованием, в котором фибриллярные белки связаны с полисахаридами и другими компонентами. Приблизительно с 60-х годов начался третий этап в изучении амилоидозов, совпавший с бурным развитием техники, в том числе и медицинской. Благодаря использованию электронной микроскопии, спектральных, иммунологических, химических, разнообразных клинических методов удалось получить много данных о природе и свойствах амилоида и его ультраструктуре. Было показано, что амилоидные отложения во многих органах человека и животных имеют сходную фибриллярную структуру: фибриллы 6–13 нм в диаметре и длиной 100 нм – 1.6 мкм. Фибрилла может состоять из двух и большего количества нитей (протофибрилл), соприкасающихся или перекручивающихся друг с другом (Shirahama & Cohen, 1967; Suzuki & Terry, 1967). С помощью рентгеноструктурного анализа и инфракрасной микроскопии показано, что для амилоидных фибрилл при всех известных вариантах амилоидоза характерна складчатая упаковка полипептидных цепей, именуемая β-складчатой структурой (Glenner et al., 1974).
2.3. Современные представления о строении и формировании амилоидных фибрилл
Амилоидные отложения состоят из фибриллярных белков связанных с полисахаридами и другими компонентами. Физико-химические особенности амилоида определяют его тинкториальные свойства, выявляемые при использовании красителя Конго красного, тиофлавина Т или S.
Таким образом, термин "амилоидоз" объединяет болезни, которые характеризуются отложением белковых масс, имеющих фибриллярную ультраструктуру и обладающих двойным лучепреломлением в поляризованном свете. Значительный прогресс в выяснении структурных свойств амилоидных фибрилл был сделан с помощью рентгеновской дифракции фибриллярного материала, выделенного из биологических тканей, а также сформированного in vitro (Blake et al., 1996; Blake & Serpell, 1996; Sunde & Blake, 1997). Эти исследования показали, что все амилоидные фибриллы имеют β-складчатую структуру с отдельными β-слоями, ориентированными параллельно главной оси фибриллы. Это означает, что белок-предшественник амилоидов, не имеющий такой структуры, подвергается молекулярным перестройкам.
На сегодняшний день известно более 20 белков, образующих амилоидные фибриллы in vivo и участвующих в патогенезе амилоидозов (таблица 2), а также белки, амилоиды которых изучены только in vitro (таблица 3) (Uversky & Fink 2004). Аβ-пептид, инсулин, лизоцим, транстиретин, амилин, хантингтин, тау-белок, α-синуклеин, миоглобин, и другие различаются между собой по аминокислотным последовательностям, вторичным и третичным структурам. Однако, несмотря на это, образованные ими амилоидные фибриллы имеют β-складчатую структуру. Эксперименты in vitro со многими белками показали, что перед образованием амилоидов структура их молекул должна претерпевать трансформацию типа «α-спираль – β-складчатость», что, как правило, требует длительной инкубации и жестких условий, несовместимых с условиями in vivo: низкие значения рН, высокие температуры, добавление ряда веществ, не присутствующих в клетке и т.п., Белки-предшественники амилоидов могут иметь β-структуру, или α-спираль или содержать обе структуры. Переход растворимой формы прионного белка в фибриллярную сопровождается уменьшением содержания α-спирали и увеличением β-структуры. Аβ-пептид при образовании амилоидных фибрилл также претерпевает трансформацию структуры от α-спирали к β-структуре. Все эти данные указывают на то, что белки, вторичная структура которых представлена α-спиралью, претерпевают трансформацию типа "α-спираль – β-структура" до или во время образования фибрилл. Однако процесс фибрилообразования не всегда требует перехода α-спирали в β-структуру. Так, белок транстиретин представляет собой тетрамер, где каждая субъединица содержит только β-структуру, а молекула α-синуклеина в нативной форме представляет собой развернутую структуру. К таким белкам можно отнести и исследуемые нами белки семейства тайтина, содержащие >90% β-cкладчатости.
Таблица 2.
Амилоидогенные белки и пептиды участвующие в патогенезе амилоидозов (см. ссылки в обзоре Uversky & Fink 2004).
|
Амилоидогенный белок |
Тип структуры |
Заболевание |
Место накопления амилоидных фибрилл |
|
β-амилоид и его пептиды |
α-спираль |
болезнь Альцгеймера |
мозг |
|
тау-белок |
развернутый |
болезнь Альцгеймера, болезнь Паркинсона |
мозг |
|
транстиретин |
β-структура |
сенильный системный амилоидоз, наследственная амилоидная полинейропатия |
во всех органах и тканях |
|
хантингтин |
α-спираль |
болезнь Хантингтона |
мозг |
|
легкие цепи иммуноглобулинов |
β-структура |
амилоидоз ассоциированный с легкими цепями |
во всех органах и тканях |
|
аполипопротеин А1 |
α-спираль |
наследственный системный амилоидоз |
глаза |
|
лизоцим |
α-спираль + β-структура |
наследственный системный амилоидоз |
внутренние органы и ткани |
|
α-синуклеин |
развернутый |
болезнь Паркинсона, деменция с тельцами Леви |
мозг |
|
амилин |
развернутый |
диабет второго типа |
печень |
|
фибриноген и его фрагменты |
β-структура |
наследственный почечный амилоидоз |
почки |
|
β2-микроглобулин |
β-структура |
амилоидоз связанный с гемодиализом |
опорно-двигательная система, сердце мочеполовая система, периферическая нервная система, желудочно-кишечный тракт |
Продолжение таблицы 2.
|
гелсолин |
α-спираль + β-структура |
наследственный системный амилоидоз |
отдельные внутренние органы и ткани |
|
кальцитонин |
развернутый |
медуллярный рак щитовидной железы |
щитовидная железа |
|
медин |
β-структура |
амилоидоз аорты |
аорта |
|
сывороточный амилоид А и его фрагменты |
α-спираль + β-структура |
АА амилоидоз |
желудок, щитовидная железа, почки |
|
цистатин С |
α-спираль + β-структура |
наследственная цистатин С амилоидная ангиопатия (болезнь кровеносных или лимфатических сосудов) |
мозг |
|
инсулин |
α-спираль |
подкожнолокализованный амилоидоз |
кожа, мышцы |
Таблица 3.
Амилоидогенные белки и пептиды, к настоящему времени не связанные с болезнями (см. ссылки в обзоре Uversky & Fink 2004).
|
Амилоидогенный белок |
Тип структуры |
Амилоидогенный белок |
Тип структуры |
|
бетабелин 15D и 16D |
β-структура |
миоглобин |
α-спираль |
|
цитохром с552 |
α-спираль |
мышечная ацилфосфатаза |
α-спираль + β-структура |
|
SH3-домен |
β-структура |
Аполипопротеин С II |
развернутый |
|
β-лактоглобулин |
β-структура |
протимозин α |
развернутый |
|
ацилфосфатаза |
α-спираль + β-структура |
метионин аминопептидаза |
α-спираль |
Процесс олигомеризации и фибриллообразования происходит при взаимодействии молекул белка за счет электростатических, водородных и гидрофобных взаимодействий с образованием димеров – начальных строительных блоков (рис. 4). Например, значительный вклад в фибриллогенез Аβ-пептида вносят гидрофобные взаимодействия. Дальше димеры олигомеризуются в тетрамеры, октамеры и т. д. с образованием протофибрилл шириной 2–3 нм и длиной до 200 нм. Эти образования накапливаются в лаг-фазе, характерной для кинетики фибриллообразования. Окончание лаг-фазы связано с образованием протофибриллами фибрилл диаметром 7–8 нм. События, происходящие в лаг-фазе, представляют большой интерес, так как именно на этой стадии с помощью микроскопа можно наблюдать кинетику фибриллогенеза, а также морфологию постепенно формирующихся агрегатов (т. е. динамику процесса) (Zerovnik, 2002). Причем, один и тот же белок может образовывать амилоидные агрегаты разной морфологии, т. е. обладать полиморфизмом, как например, Аβ(1-40)-пептид, который образует зрелые структуры разного типа (рис. 5), такие как "ветвящиеся", "спиральные" и "ленточные" (Goldsbury et al., 2000). Полиморфизм был показан и для других белков, таких как амилин (Goldsbury et al., 1997), кальцитонин (Bauer et al., 1995), инсулин (Jimenez et al., 2002).
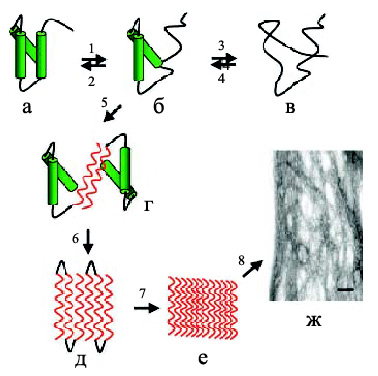
Рис. 4. Образование амилоидных фибрилл: (а) – нативная структура белка, (б) – промежуточное состояние, в котором части полипептидной цепи находятся в ненативной конформации, (в) – полностью развернутое состояние, (г) – образование межмолекулярного β-слоя, опосредованное развернутыми областями приводит к олигомеризации белка, (д) –дальнейшее образование β-складчатой структуры, (е) – образование протофибрилл, (ж) –формирование зрелых фибрилл (Jobansson, 2003).
Чемберлейн в 2000 г. показал, что фибриллы, образованные различными белками, обладают сходными структурными свойствами: все они образованы из протофибриллярных нитей, имеющих 2–5 нм в диаметре и содержащих от двух до пяти β-слоев. При этом размеры протофибрилл никак не связаны с количеством аминокислотных остатков белка-предшественника фибриллообразования. Так протофибриллы SH3 домена, включающего 90 аминокислотных остатков, состоят из двух β-слоев, а лизоцим, состоящий из 130 аминокислотных остатков, образует протофибриллярные нити, содержащие четыре β-слоя (Chamberlain et al., 2000).
2.4. Изучение амилоидных фибрилл in vitro
Первоначально, амилоидные фибриллы изучали, выделяя их из пораженных амилоидных отложений. В настоящее время для изучения амилоидных фибрилл, а также амилоидогенеза их формируют in vitro. Открытие того, что амилоидные фибриллы формируют не только белки, связанные с амилоидозами, значительно расширило эту область исследования (Dobson, 1999). Было показано, что при подходящих условиях образовывать амилоидные фибриллы in vitro могут многие белки, такие как лизоцим, миоглобин, Аβ пептид, амилин, тау-белок, хангтингтин, мышечная ацилфосфатаза и др. (Uversky & Fink 2004).
Отмечено, что амилоидогенные белки обладают различными способностями к формированию амилоидов, что отражается также в различной скорости этого процесса. Например, мышечная ацилфосфатаза человека способна формировать аморфные агрегаты после первых часов инкубации и только через 45 дней появляются пучки фибрилл (Chiti et al., 1999) (рис. 6). Аβ(1-40)-пептид после 4 часов инкубации образует аморфные агрегаты, и только после 48 часов – длинные фибриллы (Qahwash et al., 2003), а образование фибрилл α-лактальбумина занимает несколько дней (Goers et al., 2002).
За скоростью амилоидогенеза наблюдают с помощью классического амилоидного красителя тиофлавина Т, который специфически взаимодействует с амилоидными фибриллами (Krebs et al., 2005). При взаимодействии тиофлавина Т с амилоидными фибриллами происходит увеличение интенсивности флуоресценции красителя при спектрофлуорометрических измерениях и наблюдается желто-зеленая флуоресценция при флуоресцентно-микроскопических исследованиях. Молекула красителя состоит из бензтиазольного и аминобензольного колец свободно вращающихся вокруг общей С–С связи. Кребс и соавторы показали, что молекула тиофлавина Т связывается с амилоидными фибриллами специфически (Krebs et al., 2005). Они предположили, что связывание происходит в "каналах", которые тянутся вдоль β-слоев (рис. 7). Более того, тиофлавин Т связывается с амилоидными фибриллами так, что их молекулы параллельны друг другу и расположены вдоль длинной оси фибриллы (Krebs et. al., 2005). Было показано, что причиной возрастания интенсивности флуоресценции тиофлавина Т при его связывании с амилоидными фибриллами является жесткость окружения, препятствующая повороту бензтиазольного и аминобензольного колец молекулы друг относительно друга в возбужденном состоянии (Воропай и др., 2003).
Другой амилоидный краситель Конго красный также специфически взаимодействует с амилоидными фибриллами (Klunk et al., 1989). Конго красный – сульфонированый азокраситель с гидрофобной центральной частью, состоящей из бифенильной группы, расположенной между отрицательно заряженными концами молекулы красителя (рис. 8). При связывании Конго красного с амилоидными фибриллами наблюдается зеленое двойное лучепреломление в поляризационном микроскопе, и это свойство делает Конго красный наиболее используемым красителем для диагностики амилоидов. В спектральных исследованиях регистрируется сдвиг спектра поглощения Конго красного в состоянии, связанном с амилоидными фибриллами, в длинноволновую область спектра, а именно от ~490 нм к ~500 нм.
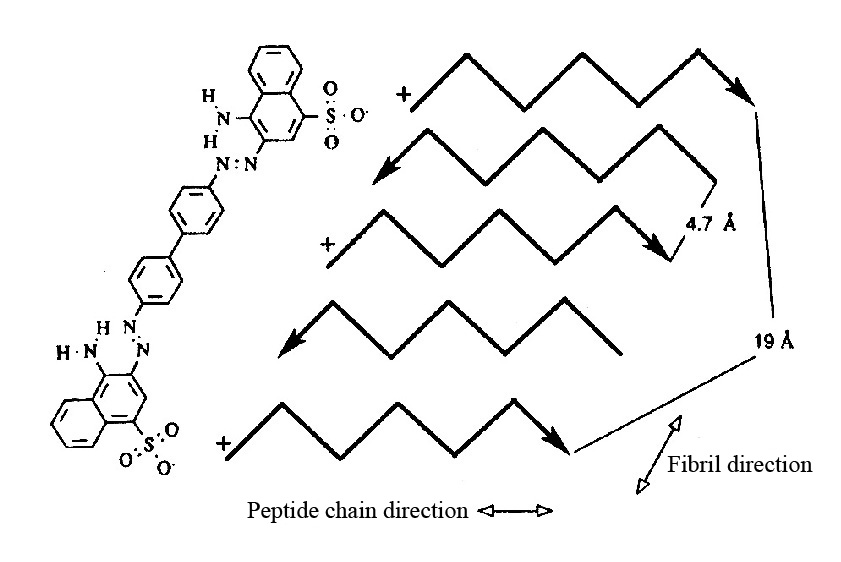
Рис. 8. Модель связывания Конго красного с амилоидными фибриллами. Показан антипараллельный слой, где каждая пятая цепочка белка имеет одинаковое N–C направление. Поэтому, молекула Конго красного может связываться с таким же типом аминокислоты в обеих полипептидных цепях (первой и пятой, как указано на рисунке). Следующая молекула красителя сможет связаться с третьей и седьмой цепочкой и т.д. (Klunk et al., 1989).
Связывание амилоидных фибрилл с Конго красным зависит от структуры амилоидных фибрилл, а именно от наличия β-складчатой структуры с отдельными β-слоями. Кланк и соавторы предложили модель связывания амилоидных фибрилл с Конго красным посредством связей между двумя отрицательно заряженными сульфоновыми группами Конго красного и двумя положительно заряженными аминокислотными остатками двух отдельных белковых молекул, которые определенным образом ориентированы в β-складчатой структуре фибрилл, образованной посредством бок о бок расположенных отдельных молекул (Klunk et al., 1989). Это означает, что белковые цепочки расположены на расстоянии 4.7 Å. Более того, каждая пятая цепочка расположена от первой на расстоянии 19 Å. Это, приблизительно, соответствует расстоянию между сульфоновыми группами КК (рис. 8). Данная модель показывает специфичность взаимодействия Конго красного с амилоидными фибриллами.
2.5. Патологические проявления амилоидозов
К настоящему времени выделяют следующие формы амилоидозов (Мягкова, 2000):
первичный (идиопатический) амилоидоз – развивается вследствие невыясненных причин;
вторичный (приобретенный) амилоидоз – развивается как осложнение после хронических заболеваний, при которых происходит распад тканей (туберкулез, бронхоэктатическая болезнь, хронический остеомиелит и др.);
наследственный (генетический, семейный) амилоидоз – врожденное нарушение белкового обмена;
старческий амилоидоз.
С другой стороны, амилоидоз разделяют на системный и локальный. Однако классификация системного амилоидоза основана на специфичности основного амилоидного белка.
Внешний вид органов при амилоидозе зависит от степени развития процесса. Если амилоидные отложения небольшие, внешний вид органа изменяется мало и амилоидоз обнаруживается лишь при гистологическом исследовании. При выраженном амилоидозе орган увеличивается в объеме, становиться очень плотным и ломким, а на срезе имеет своеобразный восковидный или сальный вид.
До сих пор остается нерешенным вопрос, касающийся механизма действия амилоидных фибрилл на органы и ткани. Исследования последних лет показали, что амилоидные агрегаты разных белков и пептидов вызывают нарушение жизнедеятельности клеток и их гибель. Как оказалось, цитотоксические свойства проявляют все амилоидные белки (Hashimoto et al., 2003; Qahwash et al., 2003; Sirangelo et al., 2004; Lee et al., 2006), однако неизвестно, чем обусловлена цитотоксичность этих агрегатов – механическим повреждением клеток, связанным с накоплением амилоидных отложений или особым молекулярным механизмом их взаимодействий внутри клетки.
цель исследования
Выяснение способности саркомерных белков семейства тайтина формировать амилоидные фибриллы in vitro.
задачи исследования
Изучить агрегационные свойства саркомерных белков семейства тайтина (тайтин, Х-, С- и Н-белки) in vitro.
Изучить агрегационные свойства Аβ(25-35)-пептида в сравнении с агрегацией молекул Х-белка.
Проверить амилоидную природу агрегатов, образуемых исследуемыми белками, поляризационной, флуоресцентной микроскопией и спектральными методами.
Изучить скорость образования амилоидных фибрилл.
Глава 3. Материалы и методы исследования
3.1. Экспериментальный и клинический материал
Экспериментальный материал: скелетные мышцы и миокард кролика.
Клинический материал: образцы миокарда пациента при ДКМП, взятые при проведении операции по пересадке сердца. Образцы миокарда человека были предоставлены ФГУ НИИ трансплантологии и искусственных органов Росздрава (г. Москва).
3.2. Выделение и очистка белковых препаратов
3.2.1. Очистка С-белка, Х-белка и Н-белка
Х-белок, С-белок и Н-белок очищали по методу (Offer et al., 1973). Фракции Х-, С- и Н-белков, полученные при хроматографической очистке миозина скелетных мышц на колонке с носителем DEAE-Sephadex А-50, концентрировали сульфатом аммония до степени насыщения 2.08 М и осаждали центрифугированием в течение 1 часа при 3000 g. Осадок растворяли в буфере, содержащем 0.3 M KCl, 4.8 мM K2HPO4, 5.2 мM KH2PO4, 0.1 мM ДТТ, 0.1 мM NaN3, pH 7.0, и диализовали против этого буфера до полного удаления сульфата аммония. Разделение белков проводили на колонке с гидроксиапатитом, уравновешенным в этом же буфере, для снятия белков с колонки использовали фосфатный градиент. Такая же процедура очистки применялась и для С-белка миокарда кролика и человека.
3.2.2. Выделение тайтина из скелетных мышц
Тайтин из скелетных мышц кролика выделяли по методу (Soteriou et al., 1993) с модификациями. Мышцы гомогенизировали в 3-х кратном (по отношению к массе мышц) объеме раствора, содержащего 50 мМ KCl, 5 мM ЭГТА, 1 мM NaHCO3,, 1 мM ДТТ, 0.1 мM NaN3, pH 7.0. Для уменьшения деградации тайтина в процессе выделения в раствор добавляли набор ингибиторов протеаз: 1 мM PMSF, 20 мкг/мл ингибитора трипсина, 10 мкг/мл леупептина.
Полученный мышечный гомогенат центрифугировали в течение 5–10 минут при 2500 g. Супернатант отбрасывали, а процедуру промывки повторяли 5–6 раз. К промытому осадку добавляли 2-х кратный объем экстрагирующего раствора, содержащего 0.9 М KCl, 2 мM MgCl2, 2 мM ЭГТА, 0.5 мM ДТТ, 0.1 мM NaN3, 1 мM PMSF, 10 мM имидазола-HCl, pH 7.0. Раствор также содержал 40 мкг/мл соевого ингибитора трипсина и 20 мкг/мл леупептина.
Экстракция проводилась на льду при 4˚С в течение 10–15 минут при непрерывном перемешивании. Экстракт осветляли в течение 40 минут при 14000 g и супернатант разбавляли в 3 раза охлажденной бидистиллированной водой, содержащей 0.1 мM ДТТ и 0.1 мM NaN3, для преципитации актомиозина (конечная ионная сила ~0.2). Через 1 час супернатант осветляли в течение 60 минут при 20000g. Для осаждения тайтина супернатант разбавляли в 5 раз (конечная ионная сила ~0.05) охлажденной бидистиллированной водой, содержащей 0.1 мM ДТТ и 0.1 мM NaN3. Через 40–60 минут осадок, содержащий преимущественно тайтин, собирали центрифугированием в течение 30 минут при 15000 g. Осадок растворяли в минимальном объеме буфера, содержащего 0.6 М KCl, 30 мM KH2PO4, 1 мM ЭГТА, 0.1 мM ДТТ, 0.1 мM NaN3, pH 7.0, и осветляли в течение 60 минут при 20000 g. Тайтин очищали методом гель-фильтрации на колонке с носителем Sepharose–CL2B, уравновешенным в этом же буфере.
3.3. Определение концентрации белковых препаратов
Концентрацию белков определяли спектрофотометрически с помощью спектрофотометра SPECOD UV VIS, используя следующие значения коэффициентов экстинции (Е2801 мг/мл): 0.543 для доколоночного миозина (Kielley & Harrington, 1960); 0.52 для колоночного миозина (Godfrey & Harrington, 1970); 1.08 для актина (Rees & Young, 1967); 1.37 для тайтина (Trinick et al., 1984); 1.09 для Х-белка, С-белка и Н-белка (Starr & Offer, 1982).
3.4. Проверка чистоты белковых препаратов
Чистоту выделенных препаратов миозина и актина скелетных мышц кролика проверяли с помощью ДСН-гель-электрофореза в 13% полиакриламидном геле по методу (Laemmli, 1970).
Чистоту тайтина, Х-белка, С-белка и Н-белка проверяли с помощью ДСН-гель-электрофореза по методу (Fritz et al., 1989) с модификациями. Согласно нашей методике, разделяющий гель содержал 7% полиакриламида вместо 15%, а также 0.75 М трис-HCl буфер, pH 8.8, 0.1% ДСН, 10% глицерина, 0.05% тетраметилэтилендиамина и 0.05% персульфата аммония. Кроме этого, вместо концентрирующего геля с содержанием полиакриламида 5% согласно (Fritz et al., 1989) применялся концентрирующий гель по методу (Laemmli et al., 1970), содержащий 2.6–2.8% полиакриламида (соотношение акриламида к бис-акриламиду 36.5:1). Эти модификации способствовали лучшему фокусированию белковых полос в геле. Концентрирующий гель также содержал 0.125 М трис-HCl буфер, pH 6.8, 0.1% ДСН, 0.05% тетраметилэтилендиамина и 0.05% персульфата аммония. Модифицированный нами метод ДСН-гель-электрофореза позволил проводить электрофоретическое разделение белков с молекулярным весом от 3 МДа до 100 кДа, что дало возможность исследовать совместное поведение тайтина, Х-белка, С-белка и Н-белка.
Электродный буфер при проведении электрофореза содержал 0.192 М глицина, 0.025 М трис и 0.1% ДСН, рН 8.3. Электрофорез проводили при токе 3–5 мА первые 30–60 минут, после этого поднимали напряжение до 12–15 мА. По окончании электрофореза гели фиксировали в растворе, содержащем 10% этанола и 10% уксусной кислоты, в течение 20–30 минут. Затем гели окрашивали в течение 30–40 минут в растворе, содержащем 0.1% кумасси G-250 и R-250 (смешанных в пропорции 1:1), 45% этанола и 10% уксусной кислоты. Отмывка окрашенных гелей проводилась в 7% уксусной кислоте при постоянном перемешивании на качалке в течение 40–50 мин.
3.5. Условия формирования амилоидных фибрилл
Амилоиды формировали диализом растворов белков в течение 20 часов при 4-37°С против растворов, содержащих: 30 мМ KCl, 10 мМ имидазола, рН 7.0; 0.15 М глицин-КОН, рН 7.0; 0.15 М глицин-KOH, рН 7.5; 25 мМ NaCI, 10 мМ Hepes, pH 7.0; 50 мM NaCl, 10 мM Hepes, pH 7.0; 30 мМ MgCI2, 10 мМ имидазола, pH 7.0; 50 мM MgCl2, 10 мM имидазола, pH 7.0.
Для исследования скорости амилоидообразования раствор Х-белка диализовали против буфера, содержащего 30 мМ KCl, 10 мМ имидазола, рН 7.0 при температуре 4˚С. Через определенные промежутки времени отбирали пробы для электронно-микроскопических и спектральных исследований. Для экспериментов по исследованию цитотоксичности амилоидных образований Х-белок также инкубировали в растворе, содержащем 30 мМ KCl, 10 мМ имидазола, рН 7.0 при температуре 4˚С.
3.6. Микроскопические исследования
3.6.1 Электронная микроскопия
Для приготовления электронно-микроскопических образцов использовали препараты белков с концентрациями 0.1 мг/мл. Каплю раствора или суспензии белка наносили на медные сетки, покрытые коллодиевой пленкой (2% коллодий в амилацетате фирмы SPI-CHEM, USA), укрепленной углеродом. После удаления избытка суспензии полоской фильтровальной бумаги образцы окрашивали 2% водным раствором уранилацетата.
Электронно-микроскопические исследования проводили на микроскопе JEM-100В при ускоряющем напряжении 80 кВ и увеличении 30000х. Увеличение микроскопа было тестировано по паракристаллам парамиозина с периодичностью 14.5 нм.
3.6.2. Поляризационная и флуоресцентная микроскопия
Исследуемые образцы, наносились на предметные стекла и высушивались на воздухе. Образовавшиеся белковые пленки окрашивались 1% водным раствором Конго красного (фирма SIGMA, USA) и затем исследовались на поляризационном микроскопе МКУ-1. Образцы также окрашивались 1% водным раствором тиофлавина Т (фирма SIGMA, USA) и исследовались с помощью люминесцентного микроскопа ЛЮМАМ-И 3.
3.7. Спектральные методы
3.7.1. Метод кругового дихроизма
Исследование вторичной структуры белка проводили методом кругового дихроизма. Спектры кругового дихроизма исследуемых белков, до и после образования ими фибрилл, регистрировали на спектрополяриметре Jasco J-600, используя кварцевые кюветы с оптической длиной 0.1 см. Спектры КД были измерены в области 200–250 нм. Обсчет спектров КД в далекой ультрафиолетовой области производился с использованием программы CONTINLL (http://lamar.colostate.edu/~sreeram/CDPro/).
3.7.2. Метод флуоресцентного анализа
Исследование амилоидной природы фибрилл, а также исследование скорости амилоидообразования было проведено с использованием классического флуоресцентного красителя амилоидных структур тиофлавина Т (ТТ). Измерения флуоресценции проводили в растворе красителя (250 мкл) и суспензии фибрилл (250 мкл) в соответствующем буфере (буфер указан в подписях под рисунками в изложении результатов). Молярное соотношение белка к красителю составляло 2:1. Интенсивность флуоресценции раствора регистрировали с помощью спектрофлуориметра PERKIN-ELMER - 44B при длине волны 488 нм и при длине волны возбуждения 420 нм. Ширина щели при возбуждении 5 нм, а при регистрации 10 нм. Из полученных значений интенсивности флуоресценции тиофлавина Т в буфере в присутствии белка вычитали значения интенсивности флуоресценции ТТ в буфере в отсутствие белка, получая интенсивность флуоресценции ТТ, связанного с белком.
3.7.3. Спектрофотометрический метод
Для подтверждения амилоидной природы фибрилл был также использован специфический краситель Конго красный (КК). Суспензию фибрилл (250 мкл) добавляли к раствору КК (250 мкл) в соответствующем буфере (буфер указан в подписях под рисунками в изложении результатов). Молярное соотношение белка к красителю составляло 2:1. Спектры поглощения КК в отсутствие и в присутствии белка регистрировали при 450–650 нм с помощью спектрофотометра SPECORD M 40.
III. Результаты и обсуждение
Глава 4. образование амилоидных фибрилл белками семейства тайтина
4.1. Электронно-микроскопическое изучение агрегационных свойств молекул тайтина, Х-белка, С-белка и Н-белка скелетных мышц кролика
Изучение амилоидогенеза разных белков in vitro проводят в условиях, которые часто не совместимы с условиями in vivo. Для образования амилоидных фибрилл белками семейства тайтина мы использовали условия, близкие к физиологическим. С помощью электронной микроскопии было показано, что исследуемые белки способны формировать разные амилоидные агрегаты, как и другие известные амилоидогенные белки (Chiti et al., 1999; Goldsbury et al., 2000; O'Nuallain et al., 2004; Uversky & Fink, 2004; см. рис. 5, 6).
Результаты электронно-микроскопических исследований представлены на рис. 9–14. Мы показали, что в растворе, содержащем 30 мМ KCl, 10 мМ имидазола, рН 7.0 Х-белок образует спирально скрученные ленточные фибриллы с осевой периодичностью ~60–70 нм, шириной ~40 нм и длиной более 1 мкм (рис. 9 А). Мы обнаружили, что такие же структуры Х-белок образует и в растворе, содержащем 0.15 М глицин-КОН, рН 7.5 (рис. 9 Б). Кроме этого Х-белок, а также С-белок и Н-белок образуют аморфные агрегаты, протофибриллы, линейные фибриллы и пучки линейных фибрилл в растворах: 50 мM NaCl, 10 мM Hepes, pH 7.0; 25 мМ NaCI, 10 мМ Hepes, pH 7.0; 50 мM MgCl2, 10 мM имидазола, pH 7.0; 30 мМ MgCI2, 10 мМ имидазола, pH 7.0; 0.15 М глицин-KOH, рН 7.5 (рис. 10–13).
На рис. 14 представлены электронные микрофотографии пучка линейных фибрилл тайтина скелетных мышц кролика в растворе 0.15 М глицин-KOH, рН 7.5. В этих условиях тайтин образует плотные пучки линейных фибрилл длиной ~3 мкм, шириной до 500 нм и аморфные агрегаты. Мы наблюдали, что аморфные агрегаты, протофибриллы длиной 100–200 нм и диаметром ~3 нм, линейные фибриллы диаметром ~7 нм и пучки линейных фибрилл длиной более 3 мкм и шириной до 500 нм могут присутствовать в одном и том же образце. Это, по-видимому, отражает разные стадии фибриллогенеза, характерные для формирования амилоидов (Kelly, 1998; Chiti et al., 1999; Goldsbury et al., 2000).
Для подтверждения амилоидной природы фибрилл тайтина, Х-белка, С-белка и Н-белка наши дальнейшие исследования были направлены на сравнение агрегатов, образуемых белками семейства тайтина, с агрегатами известных амилоидогенных белков и, в частности, Аβ-пептида, играющего важную роль в патогенезе болезни Альцгеймера.
4.2. Электронно-микроскопическое изучение агрегационных свойств Аβ(25-35)-пептида в сравнении с агрегацией молекул Х-белка
Для сравнения спирально скрученных фибрилл Х-белка с другими амилоидными фибриллами мы изучили агрегационные свойства Аβ(25-35)-пептида. Мы показали, что Аβ(25-35)-пептид, инкубированный в течение 24 ч при 37˚С образует подобно Х-белку (рис. 15 А) спирально скрученные ленты несколько микрон длиной и диаметром 25–27 нм с вариабельным осевым периодом 170–250 нм (рис. 15 Б, В).
Ленты построены из нескольких длинных и узких фибрилл диаметром 3–5 нм. На микрофотографиях они лучше видны, если смотреть вдоль ленты. Обнаруживаются также листовые агрегаты (рис. 15 Г), сформированные за счет боковой агрегации более коротких узких фибрилл. Такие агрегаты достигают в ширину ~50 нм. Таким образом, с помощью электронной микроскопии нами было установлено морфологическое сходство фибрилл Х-белка с амилоидами Aβ-пептида, найденными в мозге при болезни Альцгеймера.
4.3. Подтверждение амилоидной природы агрегатов, образуемых белками семейства тайтина (тайтина, Х-белка, С-белка и Н-белка) при их взаимодействии со специфическими красителями на амилоиды
Конго красным и тиофлавином Т
Основным методом выявления амилоидной природы фибрилл, образуемых разными белками, является их способность взаимодействовать со специфическими красителями Конго красным и тиофлавином Т (Klunk et al., 1989; LeVine, 1993, 1995; Krebs et al., 2005). Именно эти красители используют в клинической практике для определения амилоидных отложений in vivo и для исследования амилоидогенеза in vitro разными белками.
При окрашивании Конго красным фибриллярных структур, формируемых исследуемыми белками, мы наблюдали двойное лучепреломление в поляризационном микроскопе, а при окрашивании их тиофлавином Т – желто-зеленую флуоресценцию в люминесцентном микроскопе. На рис. 16 представлены данные связывания Х-фибрилл, сформированных в растворе, содержащем 30 мМ KCl, 10 мМ имидазола, рН 7.0, с красителями Конго красным и тиофлавином Т.
При спектральных исследованиях интенсивность флуоресценции тиофлавина Т в присутствии фибрилл Х-, Н- и С-белков и тайтина возрастала в ~10, ~9, ~7 и ~5 раз соответственно по сравнению с интенсивностью флуоресценции красителя в присутствии этих белков в молекулярной форме (рис. 17). Незначительное увеличение интенсивности флуоресценции тиофлавина Т наблюдается и в присутствии молекулярных форм тайтина, Х-, С- и Н-белков, что согласуется с литературными данными для белков, содержащих β-складчатую структуру (LeVine, 1993; 1995).
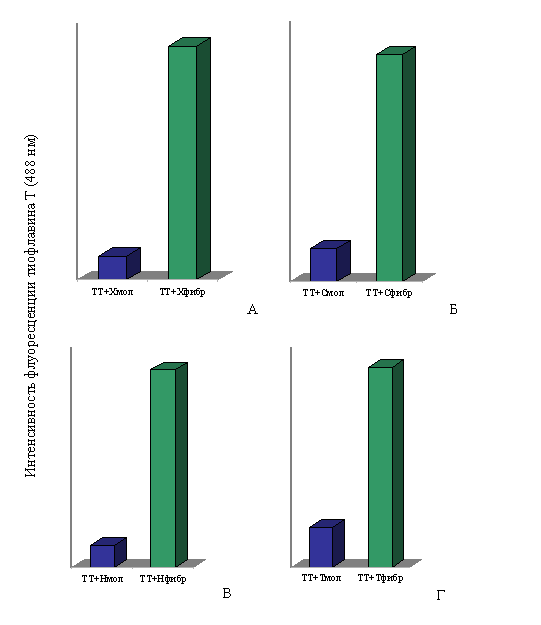
Рис. 17. Интенсивность флуоресценции тиофлавина Т (TT): А – в присутствии молекулярного X-белка (в растворе, содержащем 0.3 M KCl, 10 мМ К-фосфат, рН 7.0) и в присутствии фибрилл X-белка (0.15 М глицин-KOH, рН 7.5); Б – в присутствии молекулярного С-белка (0.3 M KCl, 10 мМ К-фосфат, рН 7.0) и в присутствии фибрилл С-белка (0.15 М глицин-KOH, рН 7.5); В – в присутствии молекулярного Н-белка (0.3 M KCl, 10 мМ К-фосфат, рН 7.0) и в присутствии фибрилл Н-белка (0.15 М глицин-KOH, рН 7.5); Г – в присутствии молекулярного тайтина (0.6 М KCl, 30 мМ KH2PO4, рН 7,0) и в присутствии фибрилл тайтина (0.15 М глицин-KOH, рН 7.5). Молярное соотношение красителя и белка 1:2.
При измерении спектральных характеристик раствора Конго красного в присутствии фибрилл тайтина, Х-, С- и Н-белков наблюдался сдвиг спектра поглощения красителя в длинноволновую область от ~490 нм к ~500 нм (рис. 18), что также является характерной чертой при связывании амилоидных фибрилл с Конго красным (Klunk et al., 1989).
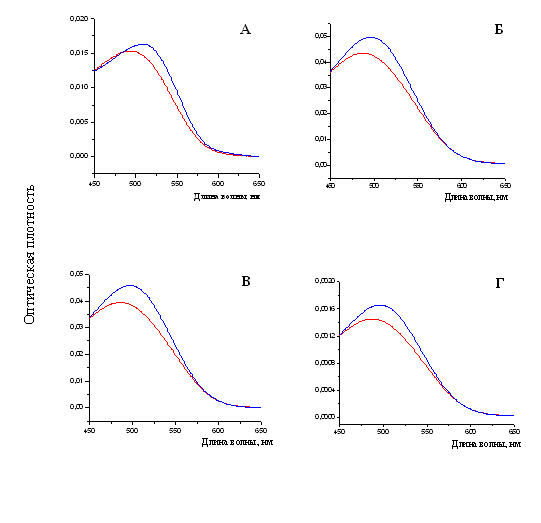
Рис. 18. Спектры поглощения свободного красителя показаны линиями красного цвета. Спектры поглощения красителя Конго красного (линия синего цвета): А – в присутствии фибрилл X-белка (в растворе, содержащем 0.15 М глицин-KOH, рН 7.5); Б – в присутствии фибрилл С-белка (0.15 М глицин-KOH, рН 7.5); В – в присутствии фибрилл Н-белка (0.15 М глицин-KOH, рН 7.5); Г – в присутствии фибрилл тайтина (0.15 М глицин-KOH, рН 7.5). Молярное соотношение красителя и белка 1:2.
Проведенные исследования указывают на специфичность связывания красителей с фибриллярными агрегатами, образуемыми белками семейства тайтина, подтверждая их амилоидную природу.
4.4. Изучение вторичной структуры тайтина и белков его семейства до и после образования амилоидных фибрилл
В работах по изучению образования амилоидных фибрилл белками in vitro используется длительная икубация и условия, несовместимые с условиями in vivo (денатурирующие вещества, низкие значения рН, высокие температуры, добавление ряда веществ, не присутствующих в клетке и т.п.) (Stine et al., 2003). Как известно, эти условия приводят к трансформации структуры молекулы белка с образованием β-складчатости, характерной для амилоидных фибрилл. (Juzczyk et al., 2005). Согласно литературным данным (Labeit & Kolmerer, 1995; Weber et al., 1993; Vaughan et al., 1993), молекулы белков семейства тайтина уже содержат β-складчатую структуру. Нами были проведены исследования вторичной структуры белков до и после образования ими амилоидных фибрилл. Как показано на рис. 20 молекулярные и фибриллярные формы Х-, С- и Н-белков характеризуются сходными спектрами кругового дихроизма (КД). Форма спектров фибрилл свидетельствует в пользу того, что в их составе практически отсутствуют α-спиральные участки, а преобладающими элементами являются β-складки. Молекулярная форма тайтина содержит ~10% α-спирали. После образования амилоидных фибрилл молекула тайтина содержит только β-структуру (рис. 19).
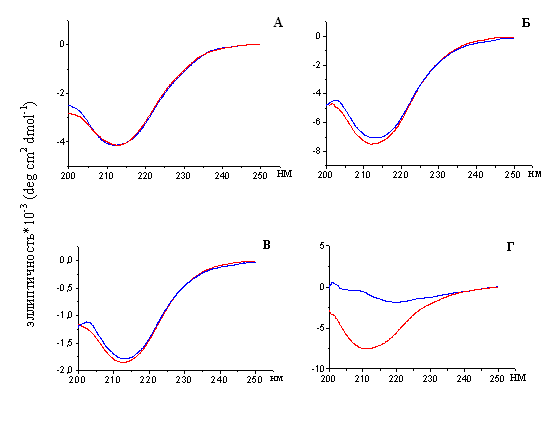
Рис. 19. Спектры кругового дихроизма (КД) в дальней Уф-области: А – Х-белка;
Б – С-белка; В – Н-белка; Г – тайтина. Спектры КД молекулярных форм Х-, С-, Н-белков (в растворе, содержащем 0.3 M KCl, 10 мМ К-фосфат, рН 7.0) и молекулярной формы тайтина (0.6 М KCl, 30 мМ KH2PO4, рН 7.0) показаны линией красного цвета. Спектры КД фибрилл Х-, С-, Н-белков и тайтина (0.15 М глицин-KOH, рН 7.5) показаны линией синего цвета.
Понятно, что при наличии у этих белков β-складчатой структуры, необходимой для образования амилоидных фибрилл, облегчается процесс формирования ими амилоидов in vitro и увеличивается опасность их быстрого роста в клетке.
4.5. Скорость образования амилоидных фибрилл Х-белка
Процессы формирования амилоидных фибрилл разными белками in vitro характеризуются разной скоростью (Kim et al., 2002; Stine et al., 2003; Hwang et al., 2004). Скорость фибриллообразования может зависеть от многих факторов: состава растворов, температуры, рН, и др. Но наибольшее влияние оказывает, скорее всего, тип вторичной структуры белка. Если белок содержит высокий процент α-спирали, то требуется трансформация типа "α-спираль – β-складчатость" (рис. 4). Так как белки семейства тайтина уже содержат β-складчатую структуру, то нет необходимости в изменении вторичной структуры. Скорость образования амилоидных структур белками семейства тайтина была продемонстрирована на примере Х-белка, так как он образует наиболее упорядоченные амилоидные структуры и методом электронной микроскопии можно визуально оценить, как происходит процесс фибриллообразования. Параллельно скорость образования амилоидных фибрилл Х-белком оценивалась по их связыванию с флуоресцентным красителем тиофлавином Т, который широко используется в качестве маркера амилоидов (LeVine, 1993).
В образцах белка происходило накопление их амилоидных структур, о чем свидетельствует возрастание флуоресцентного сигнала тиофлавина Т. Результаты представлены на рис. 20. Не происходит значительного увеличения интенсивности флуоресценции тиофлавина Т при связывании с Х-белком в первые часы инкубации (1–4 часа), так как фибриллы еще не сформированы, а присутствуют аморфные агрегаты (рис. 20 А). При дальнейшей инкубации интенсивность флуоресценции тиофлавина Т возрастала (рис. 20 Г), что соответствовало образованию амилоидных фибрилл, среди которых наблюдаются и аморфные агрегаты (рис. 20 Б). После 22 часов диализа интенсивность флуоресценции красителя при связывании с фибриллами достигала плато флуоресцентной кривой, что согласуется с данными электронной микроскопии: отмечается присутствие хорошо сформированных спирально скрученных ленточных фибрилл Х-белка (рис. 20 В). Они наблюдаются и после 26 часов диализа.
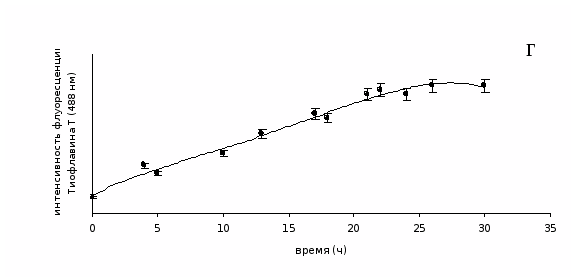
Рис. 20. А – аморфные агрегаты Х-белка (4 часа диализа), Б – линейные фибриллы и аморфные агрегаты Х-белка (17 часов диализа), В – спирально скрученные ленточные фибриллы Х-белка (22 часа диализа), сформированные в растворе, содержащем 30 мМ KCl, 10 мМ имидазола, рН 7.0. Негативное окрашивание 2% раствором уранилацетата. Шкала 100 нм. Г – скорость образования амилоидных фибрилл Х-белка.
Для многих амилоидогенных белков также характерна временная зависимость образования амилоидных фибрилл. Например, мышечная ацилфосфатаза способна формировать аморфные агрегаты после первых часов инкубации в присутствии трифлуороэтанола при рН 5.5 и температуре 25˚С, и только через 45 дней появляются пучки фибрилл (Chiti et al., 1999). Аβ(1-40)-пептид при рН 7.2, температуре 37˚С в присутствии 0.1% уксусной кислоты после 4 часов инкубации образует аморфные агрегаты и только после 48 часов – длинные фибриллы (Qahwash et al., 2003). Эксперименты со многими белками показали, что перед образованием амилоидов in vitro структура их молекул должна претерпевать трансформацию типа "α-спираль – β-складчатость", что, как правило, требует длительной инкубации и жестких условий, не совместимых с условиями in vivo (низкие значения рН, высокие температуры, добавление ряда веществ, не присутствующих в клетке и т.п.). Согласно результатам нашего исследования, образование амилоидных фибрилл Х-белком происходит в мягких условиях (рН 7.0, температура 3–5°С, ионная сила, близкая к физиологической) и быстрее, чем в случае мышечной ацилфосфатазы человека и Аβ(1-40)-пептида. Следует также отметить, что в отличие от других белков, наличие 90% β-складчатой структуры у Х-белка способствует его быстрой агрегации в амилоидные фибриллы in vitro, что указывает на возможность быстрого роста его амилоидных депозитов in vivo.
4.6. Образование амилоидных фибрилл С-белком миокарда человека при ДКМП и С-белком миокарда кролика
Амилоидные депозиты нередко обнаруживаются в сердце и кровеносных сосудах при кардиомиопатиях и миокардитах. Особо следует отметить амилоидоз сердца (кардиопатический амилоидоз или амилоидная кардиомиопатия), который, как утверждают медицинские специалисты (Сторожаков и др., 2000), к сожалению, не включается в схему дифференциального диагноза даже при резистентной к лечению сердечной недостаточности и диагностируется посмертно. Учитывая все перечисленное выше, мы решили проверить, может ли С-белок, выделенный из сердца пациента с дилатационной кардиомиопатией, образовывать амилоидные фибриллы. Дилатационная кардиомиопатия (ДКМП) – заболевание миокарда неизвестной этиологии, диагностирующееся по расширению (дилатации) левого, правого или обоих желудочков. Нарушается систолическая функция желудочков, возможно развитие застойной сердечной недостаточности, часто наблюдается нарушение ритма желудочков и предсердий. В ходе развития ДКМП сердце постепенно, но необратимо, теряет свою функциональную активность (Хубутия, 2001; Шумаков и др., 2003).
Наши исследования показали, что в разных растворах (30 мМ KCl, 10 мМ имидазола, рН 7.0; 0.01 М К-фосфат, pH 7.0; 30 мМ CaCl2, 10 мМ имидазола, рН 7.0; 30 мМ NaCl, 10 мМ имидазола, рН 7.0; 30 мМ MgCl2, 10 мМ имидазола, рН 7.0; 50 мМ MgCl2, 10 мМ имидазола, рН 7.0; 0.15 М глицин-КОН, рН 7.0) С-белок миокарда человека образует аморфные агрегаты и пучки линейных фибрилл длиной до 3 мкм и шириной до 500 нм (рис. 21 Г).
С-белок миокарда кролика в тех же условиях образует аморфные агрегаты и пучки линейных фибрилл длиной более 2 мкм и шириной до 500 нм (рис. 21 А–В). Амилоидная природа фибрилл С-белка миокарда человека и кролика была подтверждена поляризационной и флуоресцентной микроскопией, а также спектральными методами при взаимодействии их с Конго красным и тиофлавином Т (Марсагишвили и др., 2006).
Таким образом, с помощью разных специфических тестов мы показали, что белки семейства тайтина способны формировать амилоиды in vitro. Дальнейшие наши исследования должны быть направлены на тестирование токсических свойств амилоидов этих белков, на поиск подходов к их разрушению и предотвращению их образования.
СПИСОК ЛИТЕРАТУРЫ
Барсуков А., Шустов С., Шкодкин И., Воробьев С., Пронина Е. (2005) Гипертрофическая кардиомиопатия и амилоидоз сердца // Врач Вып. 10. С. 42–46.
Виноградова О.М. (1980) Первичный и генетические варианты амилоидоза // М. Медицина 224 с.
Вихлянцев И.М. (2005) Изучение тайтина и белков его семейства в скелетных мышцах в норме, при гибернации и микрогравитации // диссертационная работа. Пущино. 105 с.
Вихлянцев И.М., Макаренко И.В., Халина Я.Н., Удальцов С.Н., Малышев С.Л., Подлубная З.А. (2000) Изменения изоформного состава цитоскелетного белка тайтина – адаптационный процесс при гибернации // Биофизика. Т. 45. Вып. 5. С. 831–835.
Вихлянцев И.М., Алексеева Ю.А., Шпагина М.Д., Удальцов С.Н., Подлубная З.А. (2002) Изучение свойств С-белка скелетных и сердечных мышц сусликов Citellus undulatus на разных стадиях зимней спячки // Биофизика. Т. 47. Вып. 4. С. 701–705.
Вихлянцев И.М., Подлубная З.А., Шенкман Б.С., Козловская И.Б. (2006) Полиморфизм тайтина скелетных мышц при экстремальных условиях зимней спячки и микрогравитации: диагностическая ценность изоформ тайтина для выбора подходов к коррекции "гипогравитационного мышечного синдрома" // Докл. Акад. Наук. Т. 407. № 5. С. 692–694.
Гуровский Н.Н., Еремин А.В., Газенко О.Г., Егоров А.Д., Брянов И.И., Генин А.М. (1975) Медицинские исследования в космических полетах кораблей «Союз-12, 13, 14,» и орбитальной станции «Салют-3» // Космич. Биол. и мед. № 2. С. 48–53.
Лукоянова Н.А., Шпагина М.Д., Удальцов С.Н., Игнатьев Д.А., Колаева С.Г., Подлубная З.А (1996) Изменения в структурной организации реконструированных нитей миозина из скелетных мышц зимоспящих сусликов Citellus undulatus во время пробуждения // Биофизика. Т. 41. С. 116–122.
Макаренко И.В., Шпагина М.Д., Вишневская З.И., Подлубная З.А. (2002) Изменение структуры и функциональных свойств цитоскелетного эластичного белка тайтина при дилатационной кардиомиопатии // Биофизика. Т. 47. Вып. 4. С. 706–710.
Макаренко И.В. (2004) Роль полиморфизма тайтина в регуляции структурно-функциональных свойств миокарда в норме и при патологии // Диссертационная работа. Пущино. 107 с.
Марсагишвили Л.Г., Осипова Д.А., Вихлянцев И.М. (2006) С-белок миокарда человека образует амилоидные фибриллы // Тезисы докл. 9-ой Всероссийской медико-биологической конференции молодых ученых «Человек и его здоровье», 22 апреля, Санкт-Петербург, С. 207.
Мягкова Л.П. (2000) Энтеропатический амилоидоз: особенности клинических проявлений, место среди других форм амилоидоза. // Клиническая медицина. № 1. С. 11–14.
Подлубная З.А. (1981) Формирование сократительных структур в миогенезе // В кн.: Проблемы миогенеза. Л. с. 51–74.
Сторожаков Г.И., Гендлин Г.Е. (2000) Амилоидоз сердца // Сердечная недостаточность. Т. 1. № 1.
Фрейдина Н.А., Орлова А.А., Подлубная З.А. (1980) Электронно-микроскопическое исследование структуры С-белка и его взаимодействия с миозином, фрагментами миозина и актином // В кн.: Структурные основы и регуляция биологической подвижности. М. 160–163 с.
Хубутия М.Ш. (2001) Дилатационная кардиомиопатия // Вестник трансплантологии и искусственных органов, № 3–4. C. 32–40.
Шубникова Е.А., Юрина Н.А., Гусев Н.Б., Балезина О.П., Большакова Г.Б. (2001) Мышечные ткани // М: Медицина. 240 с.
Шумаков В.И., Хубутия М.Ш., Ильинский И.М. (2003) Дилатационная кардиомиопатия // ООО «Издательство Триада». 448 с.
Alyonycheva T.N., Mikawa T., Reinach F.C., Fischman D.A. (1997) Isoform-specific interaction of the myosin-binding proteins (MyBPs) with skeletal and cardiac myosin is a property of the C-terminal immunoglobulin domain // J Biol Chem. V. 272 (33). P. 20866–20872.
Bahler M., Moser H., Eppenberger H.M., Wallimann T. (1985) Heart C-protein is transiently expressed during skeletal muscle development in the embryo, but persists in cultured myogenic cells // Develop. Biol. V. 112. P. 345–352.
Bauer H.H., Aebi U., Haner M., Hermann R., Muller M, Merkle H.P. (1995) Architecture and polymorphism of fibrillar supramolecular assemblies prodused by in vitro aggregation of human calcitonin. // J. Struct. Biol. V. 115. P. 1–15.
Bennett P., Craig R., Starr R., Offer G. (1986) The ultrastructural location of C-protein, X-protein and H-protein in rabbit muscle // J. Muscle. Res. & Cell Motil. V. 7 (6). P. 550–567.
Bennett P., Starr R., Elliott A., Offer G. (1985) The structure of C-protein and X-protein molecules and a polymer of X-protein // J. Mol. Biol. V. 184. P. 297–309.
Blake C.C.F., Serpel L.C. (1996) Synchrotron X-ray studies suggest that the core of the transtyretin amyloid fibrils is a continuous β-sheet helix // Structure V. 4. P. 989–998.
Blake C.C.F., Serpel L.C. Sunde M., Sangren O., Lundgren E. (1996) A molecular model of the amyloid fibrils. The nature and origin of amyloid fibrils.// Ciba Found. Simp. V. 199. P. 6–21.
Callaway J.E., Bechtel P.J. (1981) C-protein from rabbit soleus (red) muscle // Biochem. J. V. 195. P. 463–469.
Chiti F., Webster P., Taddei N., Clark A., Stefani M., Ramponi G., Dobson Ch. (1999) Designing conditions for in vitro formation of protofilaments and fibrils // PNAS V. 96. P. 3590–3594.
Dobson C.M. (2001) The structural basis of protein folding and its links with human disease // Phil. Trans. Roy. Soc. Ser. B. V. 356. P. 133–145.
Draper M. H., Hodge A.J. (1949) Electron microscopy of muscle // Austr. J. Exp. Biol. Med. Sci. V. 27. P. 465–483.
Fandrich M., Fletcher M.A., Dobson S.M. (2001) Amyloid fibrils from muscle myoglobin // Nature V. 410. P. 165–166.
Flashman E., Redwood C., Moolman-Smook J., Watkins H. (2004) Cardiac myosin binding protein C: its role in physiology and disease // Circ. Res. V. 94 (10). P. 1279–1289.
Franzini-Armstrong G., Porter K.R. (1964) Sarcolemmal invaginations constituting the T-system in fish muscle tiber // J. Cell Biol. V. 22. P. 675–696.
Freiburg A., Gautel M. (1996) A molecular map of the interactions between titin and myosin binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy // Eur. J. Biochem. V. 235. P. 317–323.
Fritz J.D., Swartz D.R., Greaser M.L. (1989) Factors affecting polyacrilamide gel electrophoresis and electroblotting of high-molecular-weight myofibrillar proteins // Analyt. Biochem. V. 180. P. 205–210.
Funatsu T., Kono E., Higuchi H., Kimura S., Ishiwata S., Yoshioka T., Maruyama K., Tsukita S. (1993) Elastic filaments in situ in cardiac muscle: deep-etch replica analysis in combination with selective removal of actin and myosin filaments // J. Cell Biol. V. 120. P. 711–724.
Fürst D.O., Osborn M., Nave R., Weber K. (1988) The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line // J. Cell Biol. V. 106. P. 1563–1572.
Fürst D.O., Vinkemeier U., Weber K. (1992) Mammalian skeletal muscle C-protein: purification from bovine muscle, binding to titin and the characterization of a full-length human cDNA // J Cell Sci. V. 102. P. 769-778.
Glenner G., Eanes E., Bladen H., Linke R. (1974) β-plated sheet fibrils. A comparison of native amyloid with synthetic protein fibrils // J. Histochem. Cytochem. V. 22. P. 1141–1158.
Godfrey J.E., Harrington W.F. (1970) Self-association in the myosin system at high ionic strength. II. Evidence for the presence of a monomer-dimmer equilibrium // Biochemistry. V. 9 (4). P. 894–908.
Goedert M. (2001) α-Synuclein and neurodegenerative diseases // Nature Rev. Neurosci. V. 2. P. 492–501.
Goldsbury C.S., Wirtz S., Müller S.A., Sunderji S., Wicki P., Aebi U., Frey P. (2000) studies on the in vitro assembly of Aβ(1-40): implications for the search for Aβ fibril formation inhibitors // J. of Struct. Biol. V. 130. P. 217–231.
Gregorio C.C., Granzier H., Sorimachi H., Labeit S. (1999) Muscle assembly: a titanic achievement? // Curr. Opin. Cell Biol. V. 11. P. 18–25.
Guijarro J.I., Sunde M., Jones J.A., Campbell I.D., Dobson C.M. (1998) Amyloid fibril formation by an SH3 domain // Proc. Natl. Acad. Sci. USA V. 95. P.4224–4228.
Hanson J., O'Brien E. J., Bennett P. M. (1971) Structure of the myosin-containing filament assembly (A-segment) separated from frog skeletal muscle // J. Mol. Biol. V. 58. P. 865–871.
Hartzell H.C. (1985) Effects of phosphorylated and unphosphorylated C-protein on cardiac actomyosin ATPase //J. Moll. Biol. V. 186. P. 185–195.
Hartzell H.C., Glass D.B. (1984) Phosphorylation of purified cardiac muscle C-protein by purified cAMF-dependent and endogenous Ca2+-calmodulin-dependent protein kinases // J. Biol. Chem. V. 259. P. 15587–15596.
Hartzell H.C., Titus L. (1982) Effect of cholinergic and adrenergic agonists on phosphorylation of a 165000-dalton myofibrillar protein in intact amphibian cardiac muscle // J. Biol. Chem. V. 257. P. 2111–2120.
Hashimoto M., Rockenstein E., Crews L., Masliah E. (2003) Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases // Neuromolekular Med. V. 4. P. 21–36.
Houmeida A., Holt J., Tskhovrebova L., Trinick J. (1995) Studies of the interaction between titin and myosin // J. Cell Biol. V. 131. P. 1471–1481.
Hwang W., Zhang Sh., Kamm R.D., Karplus M. (2004) Kinetic control of dimmer structure formation in amyloid fibrillogenesis // PNAS V. 101. P. 12916–12921.
Improta S., Politou A.S., Pastore A. (1996) Immunoglobulin-like modules from titin I-band: extensible components of muscle elasticity // Structure V. 4. P. 323–337.
Itoh Y., Kimura S., Suzuki T., Ohashi K., Maruyama K. (1986) Native connectin from porcine cardiac muscle // J. Biochem. V. 100. P. 439–447.
Jeacocre S.A., England P.J. (1980) Phosphorylation of a myofibrillar protein of Mr 150000 in perfused rat heart, and the tentative identification of this as C-protein // FEBS Letters. V. 122. P. 129–132.
Juszczyk P., Kolodziejczyk A.S., Grzonka Z. (2005) Circular dichroism and aggregation studies of amyloid β (11-28) fragment and its variants // Acta Biochim. Pol. V. 52. P. 425–431.
Kelly J.W. (1998) The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways // Curr. Opin. Struct. Biol. V. 8. P. 101–106.
Kielley W.W., Harrington W.F. (1960) A model for the myosin molecule // Biochim. Biophys. Acta. V. 41 (3). P. 401–421.
Kim Y., Randolph T.W., Stevens F.J., Carpenter J.F. (2002) Kinetics and energetics of assembly, nucleation, and growth of aggregates and fibrils for an amylodogenic protein // J. Biol. Chem. V. 277. P. 27240–27246.
Klunk W.E., Pettegrew J.W., Abraham D.J. (1989) Quantitative evaluation of Congo red binding to amyloid-like proteins with a beta-pleated sheet conformation // J. Histochem. Cytochem. V. 37. P. 1273–1281.
Koretz J.F., Irving T.C., Wang K. (1993) Filamentous aggregates of native titin and binding of C-protein and AMP-desaminase // Arch. Biochem. Biophys. V. 304 (2). 305–309.
Krebs M.R., Bromley E.H., Donald A.M. (2005) The binding of thioflavin-T to amyloid fibrils: localisation and implications. // J. Struct. Biol. V. 149. P. 30–37.
Kulikovskaya I., McClellan G., Flavigny J., Carrier L., Winegrad S. (2003) Effect of MyBP-C binding to actin on contractility in heart muscle // J. Gen. Physiol. V. 122 (6). 761–774.
Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. (2000) Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2 // Circ Res. V. 86 (1). P. 51–58.
Labeit S. & Kolmerer B. (1995) Titins, giant proteins in charge of muscle ultrastructure and elasticity // Science. V. 270. P. 293–296.
Laemmli H. (1970) Clevage of structural proteins during the assembly of the head of bacterophage T4 // Nature. V. 227 (5259). P. 680–685.
Lee E.K., Park Y.W., Dong Y.Sh., Mook-Jung I., Yoo Yu. J. (2006) Cytosolic amyloid-β peptide 42 escaping from degradation induces cell death // Biochem. Biophys. Res. Communs. V. 344. P. 471–477.
LeVine III H. (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution // Prot. Sci. V. 2. P. 404–410.
LeVine III H. (1995) Thioflavine T interactions with amyloid β-sheet structures // Amyloid. V. 2. P. 1–6.
Lim M.S., Sutherland C., Walsh M.P. (1985) Phosphorylation of bovine cardiac C-protein by protein kinase C // Biochem. Biophys. Res. Communs. V. 132. P. 1187–1195.
Linke W.A., Kulke M., Li H., Fujita-Becker S., Naegoe C., Manstein D.J., Gautel M., Fernandez J.M. (2002) PEVK domain on titin: an entropic spring with actin-binding properties // J. Struct. Biol. V. 137. P. 194–205.
Liversage A.D., Holmes D., Knight P.J., Tskhovrebova L., Trinick J. (2001) Titin and the sarcomere symmetry paradox // J. Mol. Biol. V. 305. P. 401–409.
Maruyama K., Kimura S., Ohashi K., Kuwano Y. (1981) Connectin, an elastic protein of muscle. Identification of “titin” with connectin // J. Biochem. V. 89. P. 701–709.
Maruyama K., Matsub>ara R., Natori Y., Nonomura S., Kimura S., Ohashi K., Murakami F., Handa S., Eguchi G. (1977) Connectin, an elastic protein of muscle // J. Biochem. V. 82. P. 317–337.
McClellan G., Kulikovskaya I., Winegrad S. (2001) Changes in cardiac contractility related to calcium-mediated changes in phosphorylation of myosin-binding protein C // Biophys. J. V. 81 (2). P. 1083–1092.
McClellan G., Kulikovskaya I., Flavigny J., Carrier L., Winegrad S. (2004) Effect of cardiac myosin-binding protein C on stability of the thick filament // J. Mol. Cell Cardiol. V. 37 (4). P. 823–835.
Mohamed A.S., Dignam J.D., Schlender K.K. (1998) Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites // Arch. Biochem. Biophys. V. 358 (2). P. 313–319.
Moos C. (1981) Fluorescence microscope study of the binding of added C-protein to skeletal muscle myofibrils // J. Cell Biol. V. 90 P. 25–31.
Moos C., Dubin J., Mason C., Besterman J. (1976) Binding of C-protein to F-actin // Biophys. J. V. 16. P. 47a.
Moos C., Mason C.M., Besterman J. M., Feng I-N. M., Dubin J.H. (1978) The binding of skeletal muscle C-protein to F-actin and its relation to the interaction of actin with myosin sub>fragment-1 // J. Mol. Biol. V. 124. P. 571–586.
Moos C., Offer G., Starr R., Bennett P. (1975) Interaction of C-protein with myosin, myosin rod and light meromyosin // J. Mol. Biol. V. 97. P. 1–9.
Muhle-Goll C., Pastore A., Nilges M. (1998) The 3D structure of a type I module from titin: a prototype of intracellular fibronectin type III domains // Structure. V. 6. P. 1291-1302.
Nave R., Furst D.O., Weber K. (1989) Visualization of the polarity of isolated titin molecules: a single globular head on a long thin rod as the M band anchoring domain? // J. Cell Biol. V. 109. P. 2177–2187.
Offer G., Moos C., Starr R., (1973) A new protein of the thick filaments of vertebrate skeletal myofibrils. Extraction, purification and characterization // J. Mol. Biol. V. 74. P. 653–676.
O'Nuallain B., Williams A.D., Westermark P., Wetzel R. (2004) Seeding specificity in amyloid growth induced by heterologous fibrils // J. Biol. Chem. V. 279. P. 17490–17499.
Pepe F. A. (1967) The myosin filament. I. Structural organization from antibody staining observed in electron microscopy // J. Mol. Biol. V. 27. P. 203–225.
Podlubnaya Z.A., Freydina N.A., Lednev V.V. (1990) The axial repeats in paracrystals of light meromyosin and its complex with C-protein // Gen. Physiol. Biophts. V. 9. P. 301–310.
Qahwash I., Weiland K., Lu Yi., Sarver R., Kletzien R., Yan R. (2003) Identification of a mutant amyloid peptide that predominantly forms neurotoxic protofibrillar aggregates // J. Biol. Chem. V. 278. P. 23187–23195.
Rees M.K., Young M. (1967) Studies on the isolation and molecular properties of homogenous globular actin. Evidence for a single polypeptide chain structure // J. Biol. Chem. V. 242 (19). P. 4449–4458.
Safer D., Pepe F.A. (1980) Axial packing in light meromyosin paracrystals // J. Mol. Biol. V. 136. P. 343–358.
Sato N., Kawakami T., Nakayama A., Suzuki H., Kasahara H., Obitana T. (2003) A novel variant of cardiac myosin-bihding protein-C that is unable to assemble into
Shirahama T., Cohen A.S. (1967) High-resolution electron microscopic analysis of the amyloid fibril. // J. Cell. Biol. V. 33. P. 679–708.
Sipe J.D., Cohen A.S. (2000) History of the amyloid fibril. // J. Struct. Biol. V. 130. P. 88–98.
Siragelo I., Malmo C., Iannuzzi C., Mezzogiorno A., Bianco .R., Papa M., Irace G. (2004) Fibrillogenesis and cytotoxic activity of the amyloid-forming apomyoglobin mutant W7FW14F // J. Biol. Chem. V. 279. P. 13183–13189.
Soteriou A., Gamage M., Trinick J. (1993) A survey of interactions made by the giant protein titin // J. Cell Sci. V. 14. P. 119–123.
Squire J.M. (1981) The structural basis of muscular contraction // New York. London. P. 349.
Starr R. & Offer G. (1971) Polypeptide chains of intermediate molecular weight in myosin preparations // FEBS Lett. V. 15. P. 40–44.
Starr R. & Offer G. (1983) H-protein and X-protein. Two new components of the thick filaments of vertebrate skeletal muscle // J. Mol. Biol. V. 170. P. 675–698.
Starr R., Offer G. (1978) Interaction of C-protein with heavy meromyosin and sub>fragment-2 // Biochem. J. V. 171. P. 813–816.
Starr R., Offer G. (1982) Preparation of C-protein, H-protein, X-protein and phosphofructokinase // In: Methods in enzymology. New York. London. V. 85. Part B. P. 130–138.
Stine W.B., Dahlgren K.N., Krafft G.A., LaDu M.J. (2003) In vitro characterization for amyloid-β peptide oligomerization and fibrillogenesis // J. of Biol. Chem. V. 278. P. 11612–11622.
Stelzer J., Patel J., Moss R. (2006) Protein kinase A-medieted acceleration of the stretch activation responce in murine skinned myocardium is eliminated by ablation of cMyBP-C // Circ. Res. V. 13. P. 884–890.
Sunde M., Blake C.C.F. (1997) The structure of amyloid fibrils by electron microscopy and X-ray diffraction. // Adv. Prot. Chem. V. 50. P. 123–159.
Suzuki J., Kimura S., Maruyama K. (1994) Electron microscope filament lengths of connectin and its fragments // J. Biochem. V. 116. P. 406–410.
Suzuki K., Terry R.D. (1967) Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. // Acta Neuropathol. (Berl.). V. 8. P. 276–284.
Tan S.Y., Perys M.B. (1994) Histopahtology // Amyloidosis. V. 25. P. 403–414.
Trinick J., Knight P., Whiting A. (1984) Purification and properties of native titin // J. Mol. Biol. V. 180. P. 331–356.
Tskhovrebova L., Trinick J. (1997) Direct visualization of extensibility in isolated titin molecules // J. Mol. Biol. V. 265. P. 100–106.
Uversky V.N., Fink A.L. (2004) Conformational constraints for amyloid fibrillation: the importance of being unfolded // Biochim. Biophys. Acta. V. 1698. P. 131–153.
Vaughan K.T., Weber F.E., Einheber S., Fichman D.A. (1993) Molecular cloning of chiken myosin-binding protein (MyBP) H (86-kDa protein) reveals extensive homology with MyBP-C (C-protein) with conserved immunoglobulin C2 and fibronectin type III motifs. // J. Biol. Chem. V. 268. P. 3670–3676.
Wang K., McClure J., Tu A. (1979) Titin: major myofibrillar components of striated muscle // Proc.Natl Acad. Sci.USA. V. 76 (8). P. 3698–3702.
Wang K & Wright J. (1988) Architecture of the sarcomere matrix of skeletal muscle: immunoelectron microscopic evidence that suggests a set of parallel inextensible nebulin filaments anchored at the Z-line // J Cell Biol. V. 107 (6 Pt 1). P. 2199–212.
Weber F.E., Vaughan K.T., Reinach F.C., Fischman D.A. (1993) Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Differential expression, conserved domain structure and chromosome assignment // Eur J Biochem. V. 216 (2). P.661–669.
Weisberg A., Winegrad S. (1996) Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle // Proc. Natl. Acad. Sci. U S A. V. 93 (17). P. 8999–9003.
Weisberg A., Winegrad S. (1998) Relation between crossbridge structure and actomyosin ATPase activity in rat heart // Circ Res. V. 83 (1). P. 60–72
Yamamoto K. & Moos K. (1983) The C-protein of rabbit red, white, and cardiac muscles // J. Biol. Chem. V. 258 (13). P. 8395–8401.
Yamamoto K. (1984) Characterization of H-protein, a component of skeletal muscle myofibrils // J. Biol. Chem. V. 259. P. 7163–7168.
Zerovnik E. (2002) Amyloid fibril formation // Eur. J. Biochem. V. 269. P. 3362–3371.