Современные методы фармацевтического анализа
СОВРЕМЕННЫЕ МЕТОДЫ ФАРМАЦЕВТИЧЕСКОГО АНАЛИЗА
1 Специфические особенности фармацевтического анализа
Фармацевтический анализ — это наука о химической характеристике и измерении биологически активных веществ на всех этапах производства: от контроля сырья до оценки качества полученного ЛВ, изучения его стабильности, установления сроков годности и стандартизации ЛФ. Фармацевтический анализ имеет свои специфические особенности, отличающие его от других видов анализа. Эти особенности заключаются в том, что анализу подвергают вещества различной химической природы: неорганические, элементорганические, радиоактивные, органические соединения от простых алифатических до сложных природных биологически активных веществ. Чрезвычайно широк диапазон концентраций анализируемых веществ. Объектами фармацевтического анализа являются не только индивидуальные ЛВ (субстанции), но и смеси, содержащие различное число компонентов.
Способы фармацевтического анализа нуждаются в систематическом совершенствовании в связи с созданием новых Л С и непрерывным повышением требований к их качеству. Причем растут требования как к степени чистоты ЛВ, так и к количественному содержанию. Поэтому необходимо широкое использование для оценки качества ЛС не только химических, но и более чувствительных физико-химических методов.
К фармацевтическому анализу предъявляют высокие требования. Он должен быть достаточно специфичен и чувствителен, точен по отношению к нормативам, обусловленным ГФ, ФС и другой НД, выполняться в короткие промежутки времени с использованием минимальных количеств испытуемых Л С и реактивов.
Фармацевтический анализ в зависимости от поставленных задач включает различные формы контроля качества ЛС: фармакопейный анализ, постадийный контроль производства ЛВ, анализ ЛФ индивидуального изготовления, экспресс- анализ в условиях аптеки и биофармацевтический анализ.
Составной частью фармацевтического анализа является фармакопейный анализ. Он представляет собой совокупность способов исследования ЛВ и ЛФ, изложенных в Государственной фармакопее или другой нормативной документации. На основании результатов, полученных при выполнении фармакопейного анализа, делается заключение о соответствии Л С требованиям ГФ (ФС, ФСП). При отклонении от этих требований ЛС к применению не допускают.
Заключение о качестве ЛС можно сделать только на основании анализа пробы (выборки). Порядок ее отбора указан либо в частной ФС, либо в общей статье ГФ XI (вып. 2).
Выполнение фармакопейного анализа позволяет установить подлинность Л В, его чистоту, определить количественное содержание фармакологически активного вещества или ингредиентов, входящих в состав Л Ф. Несмотря на то что каждый из этих этапов имеет свою конкретную цель, их нельзя рассматривать изолированно. Они взаимосвязаны, взаимно дополняют друг друга и отражают комплексный характер оценки качества ЛС. Так, например, температура плавления, растворимость, рН среды водного раствора и т.д. являются критериями как подлинности, так и чистоты ЛВ. Указанные особенности фармакопейного анализа существенно отличают его от норм и требований к методам анализа, используемых в Государственных стандартах (ГОСТ) и технических условиях (ТУ).
В ФС описаны методики соответствующих испытаний применительно к тому или иному фармакопейному ЛС. Многие из этих методик идентичны. В целях унификации способов анализа в ГФ включены общие фармакопейные статьи (ОФС), в которых систематизированы сведения о выполнении испытаний на ряд ионов и функциональных групп, а также единых методов количественного определения. Для обобщения большого объема частных сведений по фармакопейному анализу будут рассмотрены основные критерии фармацевтического анализа и общие принципы испытаний на подлинность, чистоту и количественного определения ЛВ.
2 Критерии фармацевтического анализа
На различных этапах фармацевтического анализа в зависимости от поставленных задач имеют значение такие критерии, как избирательность, чувствительность, точность, время, затраченное на выполнение анализа, израсходованное количество анализируемого ЛВ или ЛФ.
Избирательность метода очень важна при проведении анализа смесей веществ, поскольку дает возможность получать истинные значения каждого из компонентов. Только избирательные методики анализа позволяют определять содержание основного компонента в присутствии продуктов разложения и других примесей.
Требования к точности и чувствительности фармацевтического анализа зависят от объекта и цели исследования. При испытании степени чистоты ЛВ используют методики, отличающиеся высокой чувствительностью, позволяющие устанавливать минимальное содержание примесей.
При выполнении постадийного контроля производства, а также при проведении экспресс-анализа в условиях аптеки важную роль имеет фактор времени, которое затрачивается на выполнение анализа. Для этого выбирают методы, позволяющие провести анализ в наиболее короткие промежутки времени и вместе с тем с достаточной точностью.
При количественном определении ЛВ используют метод, отличающийся избирательностью и высокой точностью. Чувствительностью метода пренебрегают, учитывая возможность выполнения анализа с большой навеской ЛВ.
Мерой чувствительности реакции является предел обнаружения. Он означает наименьшее содержание, при котором поданной методике можно обнаружить присутствие определяемого компонента с заданной доверительной вероятностью. Термин «предел обнаружения» введен вместо такого понятия, как «открываемый минимум», им пользуются также взамен термина «чувствительность». На чувствительность качественных реакций оказывают влияние такие факторы, как объемы растворов реагирующих компонентов, концентрации реактивов, рН среды, температура, продолжительность опыта. Для установления чувствительности реакций все шире используют показатель поглощения (удельный или молярный), устанавливаемый спектрофотометрическим методом. Высокой чувствительностью отличаются физико-химические методы анализа. Наиболее высокочувствительны радиохимические и масс-спектральный методы, позволяющие определять 10~8-10~9 г анализируемого вещества, полярографические и флуориметрические (10~6-10"9 г); чувствительность спек- трофотомстрических методов Ю-3-10 6, потенциометрических — Ю"2 г.
Термин «точность анализа» включает одновременно два понятия: воспроизводимость и правильность полученных результатов. Воспроизводимость характеризует рассеивание результатов анализа по сравнению со средним значением. Правильность отражает разность между действительным и найденным содержанием вещества. Точность анализа у каждого метода различна и зависит от многих факторов: калибровки измерительных приборов, точности отвешивания или отмеривания, опытности аналитика и т.д. Точность результата анализа не может быть выше, чем точность наименее точного измерения.
Так, при вычислении результатов титриметрических определений наименее точная цифра — количество миллилитров ти- транта, израсходованного на титрование. В современных бюретках в зависимости от класса их точности максимальная ошибка отмеривания около ±0,02 мл. Ошибка от натекания тоже равна ±0,02 мл. Если при указанной общей ошибке отмеривания и натекания ±0,04 мл на титрование расходуется 20 мл тшпранта, то относительная погрешность составит 0,2%. Приуменьшении навески и количества миллилитров титранта точность соответственно уменьшается. Таким образом, ти- триметрическое определение можно выполнять с относительной погрешностью +(0,2-0,3)%.
Точность титриметрических определений можно повысить, если пользоваться микробюретками, применение которых значительно уменьшает ошибки от неточного отмеривания, натекания и влияния температуры. Погрешность допускается также при взятии навески.
Отвешивание навески при выполнении анализа ЛВ осуществляют с точностью до +0,2 мг. При взятии обычной для фармакопейного анализа навески 0,5 г ЛВ и точности взвешивания ±0,2 мг относительная погрешность будет равна 0,4%. При анализе ЛФ и выполнении экспресс-анализа такая точность при отвешивании не требуется, поэтому навеску берут с точностью +(0,001-0,01) г, т.е. с предельной относительной погрешностью 0,1-1%.
При выполнении количественного определения любым химическим или физико-химическим методом могут быть допущены три группы ошибок: грубые (промахи), систематические (определенные) и случайные (неопределенные).
Грубые ошибки являются результатом просчета наблюдателя при выполнении какой-либо из операций определения или неправильно выполненных расчетов. Результаты с грубыми ошибками отбрасываются как недоброкачественные.
Систематические ошибки отражают правильность результатов анализа. Они искажают результаты измерений обычно в одну сторону (положительную или отрицательную) на некоторое постоянное значение. Причиной систематических ошибок в анализе могут быть, например, гигроскопичность ЛВ при отвешивании его навески, несовершенство измерительных и физико-химических приборов, опытность аналитика и т.д. Систематические ошибки можно частично устранить внесением поправок, калибровкой прибора и т.д.
Случайные ошибки отражают воспроизводимость результатов анализа. Они вызываются неконтролируемыми переменными.
Правильность результатов определений выражают абсолютной ошибкой и относительной ошибкой (погрешностью).
Абсолютная ошибка представляет собой разность между полученным результатом и истинным значением. Эта ошибка выражается в тех же единицах, что и определяемая величина (граммах, миллилитрах, процентах).
Относительная погрешность определения равна отношению абсолютной ошибки к истинному значению определяемой величины. Выражают относительную погрешность обычно в процентах (умножая полученную величину на 100). Относительная погрешность определений физико-химическими методами включает как точность выполнения подготовительных операций (взвешивание, отмеривание, растворение), так и точность выполнения измерений на приборе (инструментальная ошибка).
Индивидуальные вещества можно определять при анализе спектрофотометрическим методом в УФ- и видимой областях с относительной погрешностью ±(2-3)%, ИК-спектрофотометрией ±(5-12)%, газожидкостной хроматографией = (3-3,5)%, полярографией ±(2-3)%, потенциометрией ±(0,3-1)%. При анализе ЛВ в многокомпонентных смесях относительная погрешность определения этими методами возрастает примерно в два раза. Сочетание хроматографии с другими методами позволяет выполнять анализ Л В в ЛФ с относительной погрешностью ±(3-7)%.
Точность биологических методов намного ниже, чем химических и физико-химических. Относительная погрешность биологических определений достигает 20-30 и даже 50%. Для повышения точности в ГФ XI введен статистический анализ результатов биологических испытаний.
Относительная погрешность определения может быть уменьшена за счет увеличения числа параллельных измерений. Однако эти возможности имеют определенный предел. Уменьшать случайную ошибку измерений, увеличивая число опытов, целесообразно до тех пор, пока она станет меньше систематической. Обычно в фармацевтическом анализе для расчетов берут среднее из трех параллельных измерений. При статистической обработке результатов определений с целью получения достоверных результатов выполняют не менее семи параллельных измерений.
3 Физические свойства, используемые для установления подлинности лекарственных веществ
Испытание на подлинность — это подтверждение идентичности анализируемого лекарственного вещества (лекарственной формы), осуществляемое на основе требований Фармакопеи или другой НД (ФС, ФСП). Испытания выполняют физическими, химическими и физико-химическими методами. Свои особенности имеют испытания неорганических, элементорганических и органических ЛВ.
Подлинность ЛВ подтверждают показатели: описание внешнего вида, его физические свойства, физические константы и растворимость в различных растворителях. Они дают ориентировочную характеристику испытуемого ЛВ.
Физические свойства твердых ЛВ оценивают по форме кристаллов или по виду аморфного вещества, его устойчивости к свету, кислороду, содержащемуся в воздухе, гигроскопичности и степени выветривания, запаху, цвету, степени белизны. Степень белизны (по ГФ XI) Л В оценивают на спектрофотометрах СФ-18 по спектру отражения образца при его освещении белым светом. Для жидкостей устанавливают цвет, запах, летучесть, подвижность, воспламеняемость.
Температура плавления — это температура, при которой вещество переходит из твердого состояния в жидкое. По ГФ XI под температурой плавления понимают интервал температур между началом плавления (появление первой капли жидкости) и концом плавления (полным переходом вещества в жидкое состояние). Интервал между началом и концом плавления не должен превышать 2°С. Температура плавления — постоянная характеристика для индивидуального ЛВ. В присутствии даже небольшого количества примесей она изменяется, что используется для подтверждения степени чистоты Л В.
Для ЛВ, неустойчивых при нагревании, согласно требованиям ГФ XI устанавливают температуру разложения, т.е. температуру, при которой происходит резкое изменение вещества (вспенивание). Если переход вещества из твердого в жидкое состояние нечеткий, то устанавливают только температуру начала или температуру конца плавления, что оговаривается в ФС или ФСП.
В ГФ XI (вып. I, с. 17) приведены три метода определения температуры плавления. Применение того или иного метода зависит от физических свойств веществ: метод 1 и 1а применяют для легкорастираемых в порошок твердых Л В, устойчивых (метод 1) и неустойчивых (метод 1а) при нагревании; методы 2 и 3 используют для Л В, не растирающихся в порошок (жпры, воск, парафин, вазелин, смолы).
Температура затвердевания — наиболее высокая температура, при которой в течение короткого времени происходит переход ЛВ из жидкого в твердое состояние.
Температуру кипения устанавливают для жидких ЛВ. Это температура, при которой жидкость превращается в пар. Для практических целей по ГФ XI используют температурные пределы перегонки — интервал между начальной и конечной температурой кипения при нормальном атмосферном давлении 101,3 кПа (760 мм рт. ст.). Начальной считают температуру кипения, при которой в приемник перегоняются первые 5 капель жидкости, а конечной — 95% жидкости.
Плотностью называют массу единицы объема вещества (массу I см3) при стандартной температуре (обычно 20°С). Определение плотности проводят с помощью пикнометра в тех случаях, когда следует установить эту константу с точностью до 0,001, или ареометра (в случае определения плотности с точностью до 0,01). Соответствующие методики описаны в ГФ XI (в. 1, с. 24-26).
Вязкость (внутреннее трение) — свойство текучих тел (жидкостей) оказывать сопротивление перемещению одной их части относительно другой при определенной температуре. Для подтверждения качества жидких ЛВ, имеющих вязкую консистенцию, обычно определяют относительную вязкость (По™.), принимая вязкость воды за единицу. Различают также динамическую (абсолютную), удельную, приведенную, характеристическую и кинематическую вязкость. Последнюю устанавливают с помощью вискозиметра Оствальда (ГФ XI, в. 1, с. 87-94).
Растворимость — свойство газообразных, жидких и твердых веществ переходить в растворенное состояние. Растворимость в фармакопейном анализе рассматривают как свойство ЛВ растворяться в различных растворителях. Растворимость прп постоянной температуре является одной из основных характеристик, с помощью которой подтверждают доброкачественность большинства ЛВ.
Для обозначения растворимости в ГФ XI приняты условные термины, указывающие количество растворителя (мл), необходимое для растворения 1 г ЛВ. Различают очень легко растворимые (до 1 мл), легко растворимые (от 1 до 10), растворимые (от 10 до 30), умеренно растворимые (от 30 до 100), мало растворимые (от 100 до 1000), очень мало растворимые (от 1000 до 10 000), практически нерастворимые (более 10 000 мл).
Методика определения растворимости по ГФ XI (вып. 1, с. 175-176) состоит в том, что навеска ЛВ вносится в отмеренный объем растворителя и непрерывно перемешивается в случае необходимости до 10 мин. при 20 ± 2°С. Растворившимся Л В считают в том случае, если в растворе при наблюдении в проходящем свете не наблюдается частиц вещества. Отклонения от этого общего правила: образование мутных растворов, растворение более продолжительное, чем в течение 10 мин. 'такие ЛВ называют медленно растворимыми). Показатели растворимости в различных растворителях указываются в ФС. В качестве растворителей, кроме воды, используются растворы кислот и щелочей (карбонатов), а также различные органические растворители (этанол, метанол, хлороформ, эфир, ацетон, гексан, дихлорэтан, этилацетат) и масла.
Метод фазовой растворимости основан на правиле фаз Гиббса, которое устанавливает зависимость между числом фаз и числом компонентов в условиях равновесия. Суть установления фазовой растворимости состоит в последовательном прибавлении увеличивающейся массы ЛВ к постоянному объему растворителя при постоянной температуре и непрерывном встряхивании. Затем с помощью диаграмм количественно определяют массу растворенного ЛВ, устанавливая процентное содержание в нем примеси. Таким образом, метод фазовой растворимости позволяет осуществить количественную оценку степени чистоты ЛВ путем точных измерений значений растворимости. Он применим ко всем соединениям, которые образуют истинные растворы, и используется для изучения стабильности и получения очищенных (до 99,5%) образцов ЛВ.
4 Химические методы установления подлинности
Идентификация неорганических лекарственных веществ
Установление подлинности неорганических Л В основано на обнаружении с помощью химических реакций катионов и анионов, входящих в состав их молекул. С точки зрения приемов выполнения испытаний и получаемых при этом результатов можно выделить несколько общих способов.
Реакции осаждения основаны на образовании нерастворимых в воде продуктов реакции, аналитический эффект можно охарактеризовать по окраске или по растворимости осадков (в органических растворителях, кислотах, щелочах).
Эту же реакцию используют для обнаружения фосфат-ионов. Арсенат-иопы дают аналогичные результаты:


Попы калия осаждают винной кислотой:

Ионы натрия осаждают цинкуранилацетатом:

Ионы калия мож-чо осадить раствором гексанитрокобальтата (III) натрия:

Некоторые реакции осаждения можно использовать для обнаружения и катионов, и анионов.
Попы магния образуют в присутствии фосфат- и аммоний-ионов осадок фосфата магния-аммония:


Фосфат-ионы образуют с раствором молибдата аммония желтый осадок фосфор-молибдата аммония:


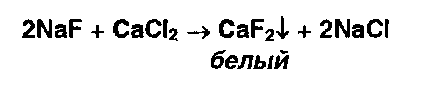




Ионы бария образуют белый осадок с сульфат-ионами: Аналогичную реакцию дают сульфиты:
Сульфит бария, в отличие от сульфата бария, растворим в хлороводородной кислоте. Ионы фтора открывают реакцией осаждения из раствора хлорида кальция:
Ионы серебра образуют осадки с хлоридами, бромидами, йодидами:
Образующиеся галогениды различаются по растворимости в растворе аммиака. Желтый осадок образуют ионы серебра с фосфатами:
Образует осадки ион серебра также с арсенит- и арсенат-ионами:
Ионы магния с растворами карбонатов образуют белый осадок основного карбоната магния:
Ионы железа (III) в растворе приобретают красное окрашивание в присутствии роданид-ионов, образуя малодис- социирующее соединение:

Ряд реактивов образуют белые или окрашенные осадки с несколькими катионами. Ионы ртути, цинка, висмута, мышьяка взаимодействуют с сульфидами:

Ионы железа (III) и цинка осаждаются растворами гсксацианоферрата (II) калия:

Ионы железа (II) дают аналогичные результаты с гексацианоферратом (III) калия:


Ионы цинка, меди и серебра осаждаются гидроксидом аммония с образованием осадков, растворимых в избытке г г-ктива:
Ионы ртути (II) и висмута (III) осаждаются йодидами, осадки растворяются в избытке реактивов:

Окислительно -восстановительные реакции, используемые для испытаний подлинности, сопровождаются изменением окраски образующихся продуктов взаимодействия.
Бромид- и йодид-ионы окисляют хлором (хлорамином, другими окислителями):

Выделившийся бром окрашивает слой хлороформа в оранжевый цвет, а йод — в фиолетовый. Йод обнаруживают также по синему окрашиванию крахмального клейстера.
Фторид-ионы
обесцвечивают красную окраску раствора
роданида железа:
 Ионы
меди, серебра
восстанавливаются из оксидов и солей
до свободных металлов:
Ионы
меди, серебра
восстанавливаются из оксидов и солей
до свободных металлов:

Нитрат- и нитрит-ионы обнаруживают путем окисления дифениламина до дифенилбензидина, а затем до дифе- нилдифенохинондиимина гидросульфата (синее окрашивание) в присутствии концентрированной серной кислоты:

Нитрит-иоиы (в отличие от нитратов) обесцвечивают раствор перманганата калия, подкисленный серной кислотой:


Взаимодействуя с антипирином (феназоном), нитриты образуют продукт замещения — нитрозоантипирин (зеленое окрашивание):

Реакции разложения сопровождаются образованием газообразных продуктов, которые обнаруживают органолеп- тически (запах, окраска).
Ионы аммония разлагаются под действием растворов гидроксидов (запах аммиака или изменение окраски красной лакмусовой бумаги):
Карбонат-ионы под действием насыщенного раствора сульфата магния образуют белый осадок, а гидрокарбонат образует осадок только при кипячении смеси (см. реакцию на магний).
Карбонат- и гидрокарбонат-ионы образуют газ — диоксид углерода под действием минеральных кислот:

Сульфит-ионы в тех же условиях образуют диоксид серы (резкий запах):
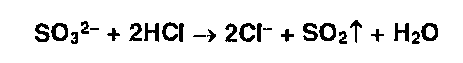
Нитрит-ионы, в отличие от нитрат-ионов, под действием кислот выделяют оксиды азота (диоксид азота имеет красно-бурую окраску):

Превращения, происходящие при нагревании и прокаливании некоторых ЛВ. Йод кристаллический, соединения мышьяка, ртути возгоняются (испытания выполнять под тягой!). Цинка оксид при прокаливании желтеет (после охлаждения окраска исчезает). Висмута нитрат основной разлагается с образованием оксида висмута (желтое окрашивание) и диоксида азота (желто-бурые пары). Соли алюминия при прокаливании с нитратом кобальта образуют плав синего цвета, представляющий собой алюминат кобальта (тенарова синь). Соли цинка в этих условиях образуют плав зеленого цвета (зелень Ринмана).
Установить наличие ряда элементов в неорганических и элементорганических ЛВ можно по изменению окраски бесцветного пламени горелки. Так, соль натрия, внесенная в пламя, окрашивает его в желтый цвет, калия — в фиолетовый, кальция — в кирпично-красный, лития — в карминово-красный. Соли бора, смоченные этанолом, окрашивают кайму пламени в зеленый цвет.
5 Идентификация элементорганических лекарственных веществ
Поскольку атомы у большинства элементорганических соединений связаны ковалентно, необходимым условием испытания их подлинности является предварительная минерализация. При этом происходит частичное или полное разрушение органической части молекулы до оксида углерода (IV) и воды. Другие элементы образуют соответствующие ионы. Последние идентифицируют с помощью рассмотренных выше или иных реакций.
Серу обнаруживают либо путем восстановления до сульфид-ионов, либо окислением до сульфат-ионов. Образование сульфида происходит также из соединений, содержащих тиоэфирную или тиокетонную серу, при нагревании с 10% раствором гидроксида натрия:

Образовавшийся при восстановлении органически связанной серы сульфид натрия идентифицируют цветной реакци- с% с н игропруссидом натрия (красно-фиолетовое окрашивание), осаждением раствором соли свинца (черное) или по сероводорода:

Счисление органически связанной серы осуществляют действием концентрированной азотной кислоты или сплавле- - .-ем со смесью нитрата и карбоната калия. Образовавшийся сульфат-ион открывают реакцией с солями бария.
Фосфорсодержащие соединения минерализуют смесью концентрированных серной и азотной кислот до фосфат-ионов, чсторые обнаруживают реакциями образования фосфата магния-аммония или фосфор-молибдата аммония (см. реакции на фосфат-ион).
Галогенсодержащие соединения под действием цинковой пыли в кислой или щелочной среде образуют галогениды:

Затем обнаруживают образовавшиеся галогенид-ионы с помощью рассмотренных выше реакций. Проба Бейльштейна основана на образовании окрашенных в зеленый цвет галогенидов меди при внесении в бесцветное пламя медной проволоки с галогенсодержащим соединением.
Фтор н хлор открывают аналитическими реакциями на соответствующие ионы после разрушения органической части молекулы расплавленным металлическим натрием:

Йод обнаруживают либо нагреванием йодпроизводного в пробирке на пламени горелки, либо действуя концентрированной серной кислотой:

Наблюдают выделение фиолетовых паров йода или фиолетовую окраску хлороформного извлечения. Можно также применить спекание со смесью нитрата калия и карбоната натрия:

Затем обнаруживают йодид-ионы.
Метод спекания можно использовать при наличии в одном соединении хлора и серы с последующим обнаружением образовавшихся хлорид- и сульфат-ионов.
Кобальт обнаруживают в виде ионов реакцией с нитрозо-Я-солью (динатриевой солью 1-нитрозо-2-нафтол-3,6-ди- . "ьфокислоты) после спекания кобальтсодержащего соединения с гидросульфитом калия (красное окрашивание).
6 Идентификация органических лекарственных веществ
Общие химические реакции
В ~армацевтическом анализе используются различные химические реакции органических соединений, которые дают определенный аналитический эффект (выпадение осадка, выделение газа, образование окрашенного раствора и т.д.).
Реаыши нитрования сопровождаются образованием окрашенных в желтый цвет моно-, ди- и тринитропроизводных арома-",'- еекого ряда:

Под действием гидроксидов калия (натрия) продукты нитрования образуют окрашенные ацисоли:



Реакции нитрозироваиия приводят к образованию окрашенных, флюоресцирующих или имеющих стабильную температуру плавления нитрозосоединений:
Фенолы образуют нитрозосоединения, бесцветные или окрашенные в сине-зеленый (фенол), сине-фиолетовый (резорцин) цвет. При нитрозировании фенолов с последующим окислением образуются индофенолы (интенсивно-синее окрашивание):
Реакции диазотирования и азосочетания используют для идентификации производных первичных ароматических аминов и фенолов. Азосоединения — окрашенные (в красный, коричневый и оранжевый цвет) продукты, получаемые в две стадии:
Диазотирование (получение соли диазония):

Азосочетание (взаимодействие соли диазония с фенолом или ароматическим амином). Сочетание происходит в ор- пю- или паря-положеннях по отношению к гидроксильной или аминогруппе, но идет легче в псрс-положении:

Аз-зс;че_гч".'.е с фенолами (нафтолами) происходит в слабощелочной (рН 9,0-10,0), а с аминами — в слабокислой сре-
Т.?: 1;: сочетания обусловлен наличием в этих соединениях электронодонорных -ОН и -ИН2 групп, создающих : -?.■■..отельные заряды в орто- и пара-положениях ароматического ядра. В этих положениях происходит электро- г ~ъг : е :гч?_;гние водорода катионом диазония и образуется азосоединение.
Г -: лзссочетания используют также для идентификации сложных эфиров фенолов, ацилированных первичных сг : • аминов (после гидролиза) и нитропроизводных (после гидрирования).
Раилии галогенирования (бромирования и йодирования) по типу реакции элеюрофильного замещения используют для обпроизводных фенолов и первичных ароматических аминов. Наличие в их молекулах заместителей первого рода (оксл- и аминогруппы) обусловливает происходящий процесс образования трибромфенола или триброманилина (белый осадок):

Аналогично происходит процесс образования трийодпроизводных. При наличии в молекулах фенола и анилина радикалов в пара- или орто-положениях образуются моно- или дигалогенпроизводные.
Реакции дегалогенирования можно выполнять без предварительной минерализации (если галогены связаны с углеродом непрочной ковалентной связью). Отщепление галогена при этом происходит под действием раствора нитрата серебра:

Дегалогенируют также, используя щелочное отщепление, путем нагревания галогенпроизводного в присутствии цинковой пыли (бромкамфора) или в спиртовом растворе гидроксида натрия:

Затем обнаруживают галогенид-ион.
Реакции конденсации альдегидов и кетонов с первичными аминами, гидроксиламином, гидразинами используются для идентификации всех указанных групп органических соединений по общей схеме:

Альдегиды, конденсируясь с первичными аминами, образуют окрашенные в желтый, красный или оранжевый цвет соли оснований Шиффа:

Эта реакция лежит в основе лигниновой пробы на первичные ароматические амины, которые взаимодействуют с ли- гнинами, содержащимися в бумаге.
Кетопроизводные образуют гидразоны:

и кетоксимы:

Гидразоны и кетоксимы — белые или окрашенные нерастворимые в воде соединения со стабильной температурой плавления. По этим признакам можно идентифицировать исходные для их получения соединения.
Окислительная конденсация с участием альдегидов лежит в основе таких широко применяемых в фармацевтическом анализе реакций, как образование ауринового красителя, нингидриновая реакция, мурексидная проба, проба Ле Розена и др.
Нингидриновая реакция является общей для а-аминокислот, иминокислот, полипептидов. Нингидрин (1,2,3-трикето- гидринденгидрат) образует с аммиаком, выделившимся из этих соединений, продукт конденсации — ион дикетогидрин- дилидендикетогидрамина, имеющий сине-фиолетовое окрашивание:

Реакции этерификации, ацилирования и гидролиза. Для подтверждения подлинности спиртов и карбоновых кислот широко используют реакцию этерификации, а подлинность сложных эфиров подтверждают с помощью обратного процесса — гидролиза:


Этерификацию проводят в присутствии дегидратирующих веществ (концентрированная серная кислота), а гидролиз — в кислой или щелочной среде.
Сходен с этерификацией процесс ацилирования (особенно ацетилирования) аминопроизводных:
а также обратный процесс — гидролиз ацильных производных.
Образовавшиеся в результате этерификации, ацилирования, гидролиза продукты идентифицируют по аналитическому эффекту (цвету, запаху, образованию газа или осадка, температуре плавления осадка и др.).
Очень широко используют, например, реакцию образования этилацетата, имеющего своеобразный фруктовый запах. Этилацетат образуют органические соединения, выделяющие при гидролизе этанол или уксусную кислоту:

Общим способом испытаний ЛВ, содержащих в молекуле сложноэфирную, лактонную, лактамную, амидную, имид- ную группы, является реакция, основанная на образовании гидроксамовых кислот (гидроксамовая проба):

Гидроксамовые кислоты, взаимодействуя с ионами железа (III) или меди (II), образуют окрашенные соли:

Реакции разложения амидов происходят при нагревании в растворах едких щелочей с образованием аммиака или алки- ламинов. имеющих характерный запах:

Первичные, вторичные и третичные амины в тех же условиях образуют соответственно метиламин, диметиламин ••" гриметиламин, например:


Указанные химические реакции используют для испытания подлинности солей первичных аммониевых оснований, амидов ароматических и гетероциклических кислот, производных уретанов.
Ациклические и циклические уреиды, алкилуреиды сульфокислот, производные гуанидина и семикарбазона, имеющие в молекуле уреидную группу, гидролизуются в щелочной среде с образованием аммиака. Например, уреиды:
К этой группе реакций можно отнести используемый в фармацевтическом анализе пиролиз (термическое разложение в сухой пробирке). Используют пиролиз для идентификации сульфаниламидов, производных бензодиазепина, пиридина и других ЛВ, которые образуют плавы с различной окраской и выделяют газообразные продукты с характерным запахом.
Реакции окисления-восстановления.
Процесс гидрирования осуществляют, как правило, водородом в момент выделения (при взаимодействии металлического цинка с хлороводородной кислотой). Эту реакцию используют для идентификации непредельных соединений, превращая их в предельные, или для восстановления нитросоединений до аминопроизводных:

Широко используются в фармацевтическом анализе реакции окисления. Первичные спирты идентифицируют, последовательно окисляя до альдегидов и кислот, которые затем обнаруживают с помощью характерных реакций:

Так, например, восстановительные свойства альдегидов устанавливают с помощью реакции образования «серебряного зеркала»:

Этот же процесс лежит в основе взаимодействия реактива Несслера с альдегидами:

Реакция окисления альдегидов лежит в основе использования реактива Фелинга, представляющего собой смесь от- лельно приготавливаемых растворов сульфата меди и калий-натриевой соли винной кислоты. В щелочной среде при нагревании в присутствии альдегидов образуется красный осадок оксида меди (I). Общая схема этой реакции:

8 Реакции образования солей и комплексных соединений
Соли органических кислот идентифицируют по наличию катионов натрия, калия, кальция и др. (с помощью рассмотренных выше реакций), а также по наличию анионов органических кислот (ацетат-, бензоат-, салицилат-, тартрат-, цитрат- и других ионов).
Широко пользуются при испытаниях на подлинность реакцией нейтрализации натриевых (калиевых) солей органических кислот (бензойной, салициловой и др.):

Нерастворимые в воде кислоты при этом осаждаются, и их идентифицируют по температуре плавления.
Нерастворимые в воде или окрашенные соли и комплексные соединения образуют с ионами тяжелых металлов органические ЛВ, содержащие в молекуле: спиртовый и фенольный гидроксил, вторичную аминогруппу, имидную группу и др. В качестве реактивов при этом используют соли железа (III), меди (II), ртути (II), кобальта, свинца, кадмия, серебра, сурьмы и др.
Ион железа (III) — наиболее широко используемый в фармацевтическом анализе реактив. Взаимодействуя с фенолами, он образует ионы феноксидов железа, окрашенные в синий, фиолетовый или красный цвет, например:

Окрашенные комплексы с ионами железа (III) образуют практически все органические соединения, содержащие в молекуле фенольный гидроксил. Если он связан в сложноэфирную группу, то реакцию выполняют после гидролиза.
Различную окраску в зависимости от рН среды имеют комплексные соединения иона железа (III) и салицилат-иона:

Структура этих комплексов обусловлена наличием у салицилат-иона не только фенольного гидроксила, но и карбоксильной группы.
Ионы железа (III) образуют окрашенные в красный цвет соли с ацетат-ионом:

а с бензоат-ионом — бензоат железа (розовато-желтый осадок):

Окнгл:е-ные комплексные соли образуют с ионами железа (III) также глюконат-, аминосалицилат-ионы, кислота ас- • : г" и тсизводные пиразолона, 8-оксихинолина, 4-оксикумарина, аминофенолы, флавоноиды и др.
Ион чети III) образует окрашенные комплексные ионы с многоатомными спиртами (глицерол, аминоспирты):


Нпичие спиртового гидроксила и вторичной аминогруппы в молекулах аминоспиртов создает условия для образования окрашенных внутрикомплексных соединений:
Аминокислоты с солями меди (II) образуют комплексные соединения, имеющие темно-синюю окраску:

Различные по растворимости и окраске внутрикомплексные соединения меди (II) образуются с сульфаниламидами. Ион меди при этом замещает подвижный атом водорода:

Подобные комплексы с амидами сульфаниловой кислоты образуют и другие ионы тяжелых металлов (кобальта, серебра).
При определенных значениях рН среды с солями меди образуют комплексные соединения барбитураты, гидроксамо- вые кислоты и др.
Ионы кальция с цитратами образуют при кипячении белый осадок цитрата кальция:

Ионы сурьмы (III) образуют окрашенные продукты с ретинолом (синий) и эргокальциферолом (оранжево-желтый).
Ионы кобальта в присутствии солей кальция образуют сине-фиолетовые комплексные соединения с барбитуратами. С производными пурина, имеющими в молекуле незамещенный атом водорода в имидной группе в положении 1 и 7, соли кобальта образуют окрашенные осадки.
Ионы серебра образуют растворимые (монозамещенные) соли с барбитуратами, производными пурина, при наличии в их молекулах незамещенной имидной группы:

Нитропруссид натрия Ыа2[Ре(СГ^)5>Ю] • НгО образует окрашенные продукты с различными органическими соединениями вследствие замещения нитрозогруппы в его молекуле, например, кетонами:

Кроме кетонов, окрашенные продукты с нитропруссидом натрия образуют альдегиды, фенолы, сульфаниламиды, производные пиридина, изоникотиновой кислоты, имидазола, ряд алкалоидов, сердечных гликозидов и др.
9 Идентификация органических оснований и их солей
Общим испытанием на соли органических оснований [Я=И] • НА с неорганическими и органическими кислотами (НА) является реакция нейтрализации связанных с ними кислот. При этом органическое основание выпадает в осадок:

Затем основание можно идентифицировать по температуре плавления или с помощью цветных реакций.
Анионы связанных неорганических (хлороводородной, бромоводородной, йодоводородной, азотной, фосфорной) и органических (бензойной, салициловой, виннокаменной и др.) кислот обнаруживают с помощью рассмотренных выше качественных реакций.
Органические азотсодержащие основания и их соли, в т.ч. алкалоиды, витамины, антибиотики, можно идентифицировать с помощью осад и тельных (общеалкалоидных) реактивов.
Осадительные реактивы образуют с органическими азотсодержащими основаниями (алифатической, ароматической, гетероциклической структуры) и их солями аморфные или кристаллические осадки (белые или окрашенные), которые имеют стабильную температуру плавления, что также подтверждает подлинность испытуемого ЛВ. Особенно широко для испытания подлинности используют пикриновую кислоту, образующую со многими органическими основаниями пикра- ты, нерастворимые в воде.
Для идентификации органических оснований и их солей используют реактивы, которые не совсем точно называют специальными (специфичными) по отношению к некоторым алкалоидам. К их числу относятся: концентрированная серная кислота, концентрированная азотная кислота, смесь этих кислот (реактив Эрдмана), концентрированная серная кислота, содержащая ванадиевую кислоту (реактив Манделина), концентрированная серная кислота, содержащая формальдегид (реактив Марки).
Концентрированная серная кислота — один из наиболее широко используемых в фармацевтическом анализе реактивов. При испытании подлинности многих органических соединений используются ее активные окислительные, дегидратирующие свойства и каталитическое действие. Сочетание концентрированной серной кислоты с другими окислителями усиливает окислительную активность этих реактивов. Кроме того, она участвует в таких химических процессах, как конденсация, кислотный гидролиз, минерализация.
Концентрированная азотная кислота используется для идентификации органических соединений, т.к. проявляет свойства окислителя и нитрующего агента. Продукты окисления приобретают различное окрашивание, а образовавшиеся ни- тропроизводные имеют характерное желтое окрашивание и легко переходят под действием гидроксидов щелочных металлов в ацисоли, имеющие хиноидную структуру и иную окраску.
10 Источники и причины недоброкачественности лекарственных веществ
Основными источниками примесей являются: исходные и промежуточные продукты синтеза, сопутствующие вещества (в растительном и животном сырье), растворители, остатки кислот и щелочей, в том числе за счет выщелачивания стекла, металл, из которого изготовлена аппаратура, песок, асбест, волокна тканей и фильтровальной бумаги и т.д.
Примеси можно разделить на две группы: технологические (внесенные исходным сырьем или образовавшиеся в процессе производства) и примеси, приобретенные в процессе хранения, транспортировки, под воздействием различных факторов (тепла, света, кислорода воздуха, влаги и др.).
Примеси могут быть токсичные (недопустимые), оказывающие влияние на фармакологический эффект, и примеси, указывающие на степень очистки ЛВ. Последние, присутствуя в больших количествах, снижают содержание биологически активных веществ и соответственно уменьшают активность ЛС. Поэтому в ФС (ФСП) указываются допустимые пределы содержания таких примесей и приводятся испытания, подтверждающие отсутствие токсичных примесей.
11 Общие требования к испытаниям на чистоту
Основное требование к испытаниям на чистоту — достаточная чувствительность, специфичность и воспроизводимость используемой реакции.
Содержание примесей можно установить эталонным и безэталонным путем. Эталонный — основан на сравнении со стандартом (эталонным раствором), содержащим определенное количество открываемой примеси. При этом в одинаковых условиях выполнения реакции наблюдают окраску или помутнение, возникающие при добавлении соответствующего реактива. Безэталонный путь — установление предела содержания примеси по отсутствию положительной реакции. При этом предел содержания примесей не превышает чувствительности реакции.
При выполнении испытаний на чистоту необходимо строго соблюдать общие указания ГФ: достаточная степень чистоты воды и растворов реактивов, точность навесок (до 0,001 г), одинаковые диаметры и цвет стекла посуды, объемы реактивов, последовательность и скорость их прибавления, единообразные условия наблюдения результатов испытаний.
12 Общие испытания на примеси неорганических ионов
Определение примесей и приблизительную оценку их количества осуществляют колориметрическим или нефеломет- рическим методами путем сравнения с эталонными растворами, нормирующими предельное содержание примеси.
Испытание иа хлориды основано на реакции с ионами серебра:

Возникает белая опалесценция, не исчезающая после добавления азотной кислоты и исчезающая при добавлении раствора аммиака:

Испытание на сульфаты основано на реакции с ионами бария:

Образуется белая опалесценция, не исчезающая от добавления хлороводородной кислоты.
Испытание на соли аммония основано на взаимодействии с реактивом Несслера:

Образуется желтое окрашивание или желто-бурый осадок.
Менее чувствителен (0,003 мг в 1 мл) способ обнаружения примеси солей аммония, основанный на выделении аммиака под действием гидроксида натрия:

Выделяющийся аммиак обнаруживают по запаху или по посинению красной лакмусовой бумаги.
Испытание на соли кальция основано на образовании белого мелкокристаллического осадка при действии оксалатом аммония:

Осадок не исчезает при добавлении уксусной кислоты, но легко растворяется в хлороводородной или азотной кислотах:



Испытание на соли железа (II) и (III) основано на образовании окрашенных феррилсульфосалицилатных солей или комплексов при взаимодействии с раствором сульфосалициловой кислоты. Окраска и состав комплексов зависят от рН среды:
В щелочных средах рН 9-11,5 образуется комплекс желтого цвета (Х>1ШХ> 416 нм), а при рН > 12 он разлагается с депрото- нированием анионного бис-комплекса:
Испытание на соли цинка основано на образовании белого осадка при взаимодействии с раствором гексацианоферрата (II) калия:

Обнаружению мешают ионы железа (III), которые в этих условиях дают синее окрашивание. Поэтому их вначале осаждают раствором аммиака.
Испытание на соли тяжелых металлов основано на образовании в уксуснокислой или нейтральной среде черного осадка или бурой окраски раствора при взаимодействии с сульфид-ионами:

13 Обнаружение примеси мышьяка
В ГФ приведено два способа определения примеси мышьяка в JIB.
Способ 1 основан на реакции Зангера-Блека и осуществляется путем восстановления соединений мышьяка (III) цинком (в присутствии хлороводородной кислоты) в специальном приборе до арсина:

Арсин, проходя через слой ваты, пропитанной ацетатом свинца, освобождается от возможной примеси сероводорода:
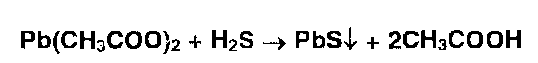
Предварительно дихлорид олова восстанавливает соединения мышьяка (V) до (III):

Затем арсин, соприкасаясь с бумагой, пропитанной раствором дихлорида ртути, окрашивает ее в зависимости от концентрации мышьяка в оранжевый или желтый цвет. Последовательно происходят реакции:
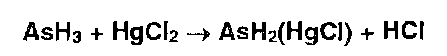

Повысить предел чувствительности реакций с 0,001 мг до 0,0005 мг (0,5 мкг) можно, если обработать бумагу раствором йодида калил. Происходит взаимодействие дихлорида ртути (I) с йодидом калия:

Окраска усиливается за счет образования металлической ртути:

Недостаток способа 1 состоит в невозможности обнаружения примеси мышьяка в присутствии соединений сурьмы, фосфора, солей тяжелых металлов, сульфид- и сульфит-ионов.
Способ 2, основанный на реакции Буго-Тиле, не имеет этого недостатка, но реакция менее чувствительна (0,01 мг). Сущность реакции обусловлена восстановительными свойствами натриевой соли фосфорноватистой кислоты (гипофо- сфита натрия), которая восстанавливает соединения мышьяка, окисляясь при этом до фосфористой кислоты. В зависимости от содержания примеси соединений мышьяка появляется бурое окрашивание или бурый осадок:

Испытание выполняют в пробирке, в которую вносят навеску испытуемого ЛВ, реактив и нагревают в кипящей водяной бане 15 мин. После охлаждения прибавляют 3 мл воды, 5 мл эфира, тщательно взбалтывают. При наличии примеси соединений мышьяка на границе слоев жидкостей образуется бурая пленка (осадок мышьяка). Этот способ применим также для определения селена и теллура.
14 Определение воды и летучих веществ
В ГФ включены два физических метода (высушивания и дистилляции) и один химический (акваметрия) метод определения воды. Метод высушивания заключается в установлении разности массы ЛВ или лекарственного сырья до и после высушивания (условия высушивания, температура и навеска указываются в ФС). Открытый бюкс вместе с крышкой охлаждают в эксикаторе в течение 50 мин и взвешивают.
Второй метод определения воды основан на свойстве двух несмешивающихся жидкостей (например, воды и толуола) перегоняться при более низкой температуре, чем каждая из этих жидкостей. Определение выполняют в специальном приборе. Затем содержание воды устанавливают по ее объему в приемнике после окончания перегонки и охлаждения.
Акваметрия, или метод титрования реактивом Фишера, состоит в определении в ЛВ как гигроскопической, так и кристаллизационной воды реактивом, включающим раствор диоксида серы, йода и пиридина в метаноле. Определение выполняют в закрытой системе, чтобы исключить влияние атмосферной влаги. Взаимодействие реактива с водой протекает по схеме:

Недостатком метода, кроме необходимости соблюдения герметичности, является невозможность его использования в присутствии веществ, реагирующих с компонентами реактива (альдегиды, кетоны, меркаптаны, сульфиды, оксиды, ги- дроксиды, карбонаты металлов и др.).
Установление рИ среды
Величина рН дает важную информацию о степени чистоты ЛВ (содержании в нем примесей кислотного и основного характера). В ряде ФС рекомендуется устанавливать кислотность или щелочность путем нейтрализации примесей кислот или щелочей в водном растворе или экстракте. Нейтрализацию проводят в присутствии индикаторов (фенолфталеин, ти- молфталеин, метиловый красный и др.). О кислотности или щелочности судят либо по окраске индикатора, либо по ее изменению, либо по количеству кислоты или щелочи, затраченных на нейтрализацию.
В большинстве случаев ФС (ФСП) регламентирует величину рН среды раствора. Это значение, ориентировочно до 0,3 единицы рН, можно установить с помощью индикаторной бумаги или универсального индикатора. Более объективные результаты дают колориметрический и потенциометрический способы. Для колориметрического определения готовят серию буферных растворов, отличающихся друг от друга на величину рН, равную 0,2. К каждому из них добавляют по 2-3 капли индикатора. Аналогично поступают с испытуемым раствором, который готовят в тех же условиях. Затем сравнивают его окраску с приготовленной серией буферных растворов и устанавливают рН среды.
Потенциометрическое определение рН выполняют на потенциометрах или рН-метрах различных марок после предварительной их настройки с помощью буферных растворов. Этот метод отличается более высокой точностью, имеет меньше ограничений, может быть применен в присутствии окислителей, восстановителей, в окрашенных и мутных растворах.
15 Испытания на чистоту по физическим и химическим свойствам
Прозрачность и степень мутности. Прозрачными считают растворы, при освещении которых шаровой электролампой (40 Вт) на черном фоне не наблюдается присутствие нерастворенных частиц. Степень мутности устанавливают путем сравнения в одинаковых пробирках растворов испытуемого вещества с растворителем или с эталонами. Эталонами служат взвеси в воде, полученные смешиванием определенных количеств 1% раствора гидразина сульфата и 10% раствора гекса- метилентетрамина.
Окраску жидкостей по ГФ XI устанавливают, сравнивая испытуемые растворы с равным количеством одного из семи эталонов при дневном освещении на матово-белом фоне. Эталоны готовят, смешивая в различных соотношениях четыре основных раствора, получаемых из исходных растворов хлорида кобальта, дихромата калия, сульфата меди (И) и хлорида железа (III). Растворителем служит раствор серной кислоты (0,1 моль/л). Бесцветными считают растворы, цвет которых не отличается от воды.
Адсорбционную способность и дисперсность устанавливают в соответствии с требованиями ФС (ФСП), Дисперсность можно установить по скорости осаждения водной суспензии испытуемого ЛВ в мерном цилиндре. Адсорбционную способность — по обесцвечиванию окраски индикатора (метиленового синего) в растворе ЛВ с определенной концентрацией и в определенном объеме.
Примесь органических веществ обнаруживают действием концентрированной серной кислоты. При этом образуются окрашенные продукты, интенсивность окраски которых не должна превышать соответствующий эталон цветности.
Примесь восстанавливающих веществ в ЛВ устанавливают по обесцвечиванию растворов перманганата калия (определенного объема и концентрации).
Примесь окрашенных веществ определяют по бесцветности водного извлечения. Обнаруживают также примесь водорастворимых солей в Л В, нерастворимых в воде, и примеси, нерастворимые в воде, в водорастворимых ЛВ (по эталону мутности).
16 Определение золы
Общую золу устанавливают прокаливанием навески ЛВ в фарфоровом (платиновом) тигле при слабом красном калении (около 500°С) до постоянной массы. После окончания прокаливания тигель охлаждают в эксикаторе и взвешивают. При последующем добавлении к остатку 15 мл 10% раствора хлороводородной кислоты и нагревании в течение 10 мин на кипящей водяной бане отфильтрованный осадок вновь сжигают, прокаливают, охлаждают и взвешивают, определяя содержание золы, нерастворимой в хлороводородной кислоте.
Сульфатную золу определяют после нагревания и прокаливания навески ЛВ, смоченной 1 мл концентрированной серной кислоты. Нагревают осторожно на сетке или песчаной бане до удаления паров серной кислоты, а затем прокаливают до постоянной массы, которую устанавливают, охлаждая в эксикаторе и взвешивая тигель. Во многих ФС (ФСП) предус мотрено последующее определение в сульфатной золе примесей тяжелых металлов.
17 Испытание на специфические примеси
К числу специфических примесей относят присущие только определенному ЛВ исходные или промежуточные продукты синтеза, продукты разложения, сопутствующие биологически активные вещества (алкалоиды, гормоны, белки, полисахариды и др.). Они могут влиять на фармакологический эффект или оказывать токсическое действие. Несмотря на многообразие химической структуры специфических примесей, для их обнаружения используют несколько основных способов:
Использование специфической примеси в качестве эталона и фотометрическое или нефелометрическое определение ее содержания по отношению к этому эталону.
Использование способов, основанных на избирательном взаимодействии примеси с каким-либо реактивом и последующем ее определении.
Экстрагирование (отделение) примеси с помощью несмешивающихся растворителей (вода-эфир), отгонка растворителя и гравиметрическое, титриметрическое или фотометрическое ее определение.
Разделение и исследование примесей с помощью хроматографических методов (ТСХ, бумажная хроматография).
Испытания на чистоту, основанные на использовании ВЭЖХ, ГЖХ и их сочетания с другими методами (спектрофо- тометрия, масс-спектроскопия, полярография).
б.б. Химические методы определения лекарственных веществ
Методики, основанные на использовании химических методов, включены в ОФС и ФС (ФСП). Наиболее широко для количественного определения ЛВ применяют титриметрическис методы анализа. Значительно реже используют гравиметрический метод, газометрический метод и элементный анализ.
Гравиметрический метод основан на измерении массы вещества. Сущность определения состоит в последовательном выполнении реакции осаждения, отделении, высушивании и взвешивании осадка. ГФ рекомендует гравиметрию для количественного определения барбитуратов, солей хинина, других ЛВ в виде органических оснований или нерастворимых в воде продуктов реакции (пикратов, кремниевольфраматов и др.).
Газометрический метод основан на взаимодействии испытуемого ЛВ с поглотительным раствором, содержащим количественно реагирующие с ним компоненты. Применяют для определения газообразных ЛВ (кислород, оксид азота, циклопропан и др.).
18 Осадительное титрование
Аргеитометрия основана на реакциях осаждения галогенидов (Hal-) титрованным раствором нитрата серебра:

При прямом аргентометрическом титровании используют индикатор хромат калия (метод Мора) или адсорбционные индикаторы (метод Фаянса), При обратном титровании (метод Фольгарда) индикатором служат жслезоаммониевые квасцы, а избыток нитрата серебра определяют роданометрическим (тиоцианатометрическим) методом.
Ъюцианатометрия основана на реакции осаждения иона серебра тиоцианатом аммония (индикатор — железоаммоние- вые квасцы):

Меркуриметрия основана на образовании малодиссоциированных соединений ртути (II):

При титровании хлоридов индикатором служит дифенилкарбазид или дифенилкарбазон:

При титровании йодидов конечную точку титрования устанавливают по выпадению красного осадка йодида ртути (II) вследствие разрушения образующегося при титровании тетрайодомеркурат-иона:



Меркурометрия — метод определения галогенидов, образующих малорастворимые соединения с катионами ртути (I). Титрантом служит раствор Ь^г^Оз):, индикатор — тиоцианат железа, который обесцвечивается в точке эквивалентности вследствие образования тиоцианата ртути (I):
19 Кислотно-основное титрование (метод нейтрализации)
Титрование в водной среде
Ациднметрия используется для определения натриевых (калиевых) солей неорганических и органических кислот, а также органических оснований (ИзМ). Титрант— раствор хлороводородной кислоты:
Алкалиметрия используется для определения неорганических и органических кислот, а также солей органических оснований с различными кислотами:

Косвенная (заместительная) нейтрализация основана на реакции осаждения ионами серебра органических оснований, содержащих в молекуле вторичную аминогруппу или меркаптогруппу:

Выделившуюся кислоту титруют алкалиметрическим методом.
Оксимный метод также основан на косвенной нейтрализации эквивалентного количества хлороводородной кислоты, выделившейся при взаимодействии гидроксиламина гидрохлорида с кетопроизводными:

Этерификация в сочетании с алкалиметрией используется при определении спиртов и фенолов. Их ацетилируют уксусным ангидридом, а его избыток гидролизуют до уксусной кислоты, которую затем оттитровывают раствором гидроксида натрия:

Параллельно выполняют контрольный опыт с тем же количеством уксусного ангидрида.
Гидролиз сложных эфиров в сочетании с ацидиметрией. Сложные эфиры гидролизуют титрованным раствором гидроксида натрия, избыток которого титруют хлороводородной кислотой:

Пиролиз может быть выполнен в кислой среде:

Образовавшуюся при гидролизе органическую кислоту можно извлечь эфиром и оттитровать алкалиметрическим методом.
20 Титрование в смешанных растворителях
Используют в тех случаях, когда ЛВ плохо растворяются в воде или водные растворы имеют слабо выраженные кислотные (щелочные) свойства. Они усиливаются в присутствии этанола (ацетона).
Титрование в воде в присутствии несмешивающихся с ней эфира или хлороформа используют для извлечения органического основания или кислоты из водной фазы, что исключает их влияние на результаты титрования.
Титрование н среде неводных растворителей (неводное титрование)
Метод позволяет количественно определить органические вещества, проявляющие в водной среде очень слабые основные или кислотные свойства. В качестве титрантов используют растворы сильных кислот или сильных оснований.
Неводное титрование органических оснований (Яз!4!) и их солей (ЯзЫ ■ НА) выполняют, используя в качестве растворителей безводные уксусную кислоту, уксусный ангидрид, муравьиную кислоту или их сочетания. Титрантом служит раствор хлорной кислоты, индикаторами — кристаллический фиолетовый, тропсолин 00, метиловый оранжевый.
Титрование слабых органических оснований хлорной кислотой в среде ледяной уксусной кислоты включает несколько этапов:
Растворение НСЮ4 в ледяной СНзСООН:

Растворение основания (Яз1Ч) в ледяной СНзСООН:

Взаимодействие ацетоний- и ацетат-ионов:

Взаимодействие протонированного амина с хлорат-ионом:

Очень слабые органические основания (рК> 12) необходимо титровать хлорной кислотой в среде уксусного ангидрида £УА), т.к. он более активно (чем ледяная уксусная кислота) усиливает основные свойства аминов.
Взаимодействие НСЮ4 с УА:

Растворение амина (ЯзИ) в УА:

Взаимодействие кислоты с основанием:
4. Взаимодействие ацетилий-иона с хлорат-ионом:

Соли органических оснований с галогеноводородными кислотами (ЯзЫ • НХ) проявляют кислотные свойства даже в неводной среде. Поэтому их титруют в присутствии ацетата ртути (II), который нейтрализует галогенпроизводную кислоту. Малодиссоциированные галогениды ртути (HgX2) и (СНзСОО)г^ не мешают определению. Образующийся ацетат органического основания оттитровывают хлорной кислотой:


Неводное титрование галогеноводородов может быть выполнено без добавления ацетата ртути, если в качестве прото- генных растворителей использовать безводную муравьиную кислоту в присутствии уксусного ангидрида.
Неводное титрование органических веществ, проявляющих кислотные свойства (фенолы, барбитураты, карбоновые кислоты, сульфаниламиды и др.), выполняют, используя в качестве растворителя диметилформамид или его смесь с бензолом. Титрантом служит раствор гидроксида натрия в смеси метанола и бензола или раствор метилата натрия. В качестве индикатора используют тимоловый синий.
Суммарно процесс нейтрализации фенолов (енолов) можно представить так:

21 Окислительно-восстановительное титрование
Йодометрия — метод, основанный на окислительных свойствах йода и восстановительных свойствах йодид-ионов:

Титрант — раствор йода (индикатор — крахмал) используют для прямого титрования неорганических и органических веществ, способных окисляться или образовывать с йодом продукты присоединения или замещения. Используют также обратное йодометрическое титрование. При этом избыток йода титруют 0,1 М раствором тиосульфата натрия:

Восстановительные свойства йодида калия используют для количественного определения веществ, обладающих окислительными свойствами. Выделившееся эквивалентное количество йода оттитровывают тиосульфатом натрия.
Используют также сочетание реакции замещения (получение нерастворимых в воде моно-, ди- и трийодпроизводных) и обратной йодометрии. Йодпроизводные отфильтровывают, а в фильтрате определяют избыток титрованного раствора йода. Аналогичным образом используют реакцию образования полийодидов [ЯзЫ] • Н1 • и органических оснований. При выполнении определения необходимо учитывать влияние рН среды.
Йодхлорометрия — отличается от йодометрии использованием в качестве титранта не раствора йода, а более устойчивого раствора йодмонохлорида. Аналогично йоду йодмонохлорид образует йодпроизводные органических оснований. Избыток титранта устанавливают йодометрически:

Йодатометрия основана на окислении органических соединений йодатом калия. Избыток титранта устанавливают йодометрически:

Брочаточетрия (бромид-броматометрия) основана на использовании окислительных свойств или реакции замещения -р.-^гкиг моно-.ди- или трибромпроизводных) за счет образующегося свободного брома:


Индикаторами при прямом титровании служат азокрасители, которые обесцвечиваются бромом в эквивалентной точ- • е метиловый красный). В случае обратного титрования эквивалентную точку устанавливают йодометрически по избыт- ?г. -'.гранта (бромата калия).
Перманганатометрия основана на использовании окислительных свойств титранта — перманганата калия в кислой среде:
Индикатором при прямом титровании служит сам титрант (появляется фиолетовое окрашивание), а при обратном титровании избыток титранта устанавливают йодометрическим методом.
Цериметрия основана на использовании окислительных свойств титранта — соли церия (IV), который в кислой среде восстанавливается до церия (III):

Индикатором служат дифениламин, о-фенантролин, а при обратном титровании избыток титранта устанавливают йодометрически:

22 Комплексономе трия
Метод основан на образовании прочных, растворимых в воде комплексов катионов металлов с трилоном Б — динатри- евой солью этилендиаминтетрауксусной кислоты (ЭДТАЫаг) или другими комплексонами. Независимо от заряда катиона взаимодействие его с титрантом (ЭДТА №2) происходит в стехиометрическом соотношении 1:1:
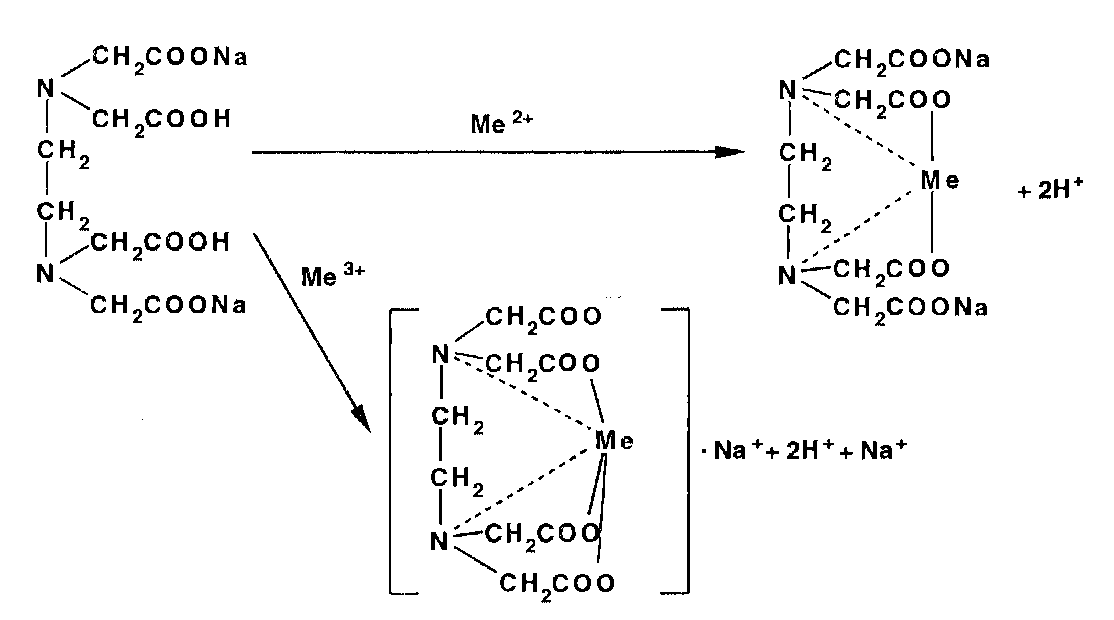
Используют метод для количественного определения неорганических и органических ЛВ, содержащих катионы магния, кальция, цинка, висмута, свинца, алюминия и др. Точку эквивалентности устанавливают с помощью металлоинди- каторов — органических красителей (ксиленоловый оранжевый, пирокатехиновый фиолетовый, кислотный хром темно-синий). образующих с указанными катионами непрочные, ярко окрашенные комплексы. В эквивалентной точке эти комплексы разрушаются до образования свободного индикатора, по окраске которого делают заключение о конце титрования.
Непременным условием комплексонометрии является строгое соблюдение при титровании определенного интервала рН, что достигается с помощью буферных растворов. Комплексонометрическое титрование может быть выполнено прямым, обратным и косвенным (заместительным) методами.
23 Нитритометрия
Метод количественного определения первичных и вторичных ароматических аминов, основанный на использовании титранта — раствора нитрита натрия в присутствии бромида натрия:




В этих условиях с первичными ароматическими аминами образуются диазосоединения (в кислой среде):
Вторичные ароматические амины в тех же условиях образуют Ы-нитрозосоединения:
Эквивалентную точку устанавливают различными путями: потенциометрически, с помощью указанных в ФС внутренних индикаторов (тропеолин 00, нейтральный красный), с внешним индикатором (йодкрахмальная бумага). Титрование с йодкрахмальной бумагой ведут до тех пор, пока капля титруемого раствора, взятая через 1 мин после прибавления титранта, не вызовет тотчас же посинение бумаги:
На результаты определения влияют температура (смесь охлаждают до 5-15°С), концентрация хлороводородной кислоты, природа растворителя.
При использовании внутренних индикаторов наблюдают изменение их окраски в эквивалентной точке.
24 Элементный анализ
Определение азота в органических соединениях (метод Кьельдаля)
Метод основан на предварительной минерализации азотсодержащего органического соединения до гидросульфата аммония. Определение выполняют с помощью прибора, состоящего из колбы Кьельдаля, парообразователя, холодильника, приемника. Оно состоит из нескольких стадий.
Минерализация (нагревание с конц. Н2504):

Разложение (1ЧН4)Н304 гидроксидом натрия и отгонка образующегося аммиака в приемник:

Взаимодействие ЫНз в приемнике с борной кислотой с образованием тетрагидроксибората аммония:

Титрование отгона ОДМ раствором хлороводородной кислоты:

Параллельно выполняют контрольный опыт (без анализируемого вещества) для повышения точности результатов анализа.
Для определения веществ, содержащих в молекуле амидную группу, используют упрощенный вариант метода Кьельдаля, исключающий стадию минерализации. Методика сводится к гидролизу амида в колбе Кьельдаля 30% раствором гид- роксида натрия, отгонке выделяющегося аммиака или амина в приемник и титровании отгона 0,1 М хлороводородной кислотой.
25 Метод сжигания в колбе с кислородом
Используется для анализа ЛВ, содержащих в молекуле галогены, серу, фосфор. Сжигание проводят в колбе из термо- ^ никого стекла, наполненной кислородом. В пробку колбы впаяна платиновая или нихромовая проволока, заканчиваются спиралью (держатель), в которую помещают точную навеску ЛВ, завернутую в фильтровальную бумагу. На дно колбы вливают поглощающую жидкость. По окончании сжигания колбу оставляют на 30-60 мин, периодически перемеши- г:- После этого химическим или физико-химическим методом идентифицируют или определяют образовавшиеся ионы.
Так. например, йодсодержащие органические соединения последовательно количественно превращают в йодаты.
Сжигание ЛВ в атмосфере кислорода приводит к окислению до свободного йода, растворяющегося в растворе гид- :: ксида натрия (поглощающая жидкость) с образованием йодида и гипойодита натрия:

Для окисления образовавшихся йодидов до йодатов в колбу вносят раствор ацетата брома до появления желтого окрашивания:

Для удаления избытка брома добавляют концентрированную муравьиную кислоту до обесцвечивания раствора:

Выдерживают 5 мин в темном месте после добавления йодида калия и раствора серной кислоты, а затем титруют выделившийся йод, содержание которого эквивалентно его количеству в испытуемом ЛВ:

26 Физические и физико-химические методы анализа
Физические и физико-химические методы могут быть классифицированы на следующие группы: оптические методы, методы, основанные на поглощении электромагнитного излучения, методы, основанные на испускании излучения, методы, основанные на использовании магнитного поля, электрохимические методы, термические методы, методы разделения.
Физико-химические методы основаны на использовании зависимости физических свойств от химического состава веществ. В большинстве случаев физико-химические методы отличаются быстротой выполнения, избирательностью, высокой чувствительностью, возможностью унификации и автоматизации. Поэтому данная группа методов приобретает все большее значение для объективной оценки качества ЛС, в т.ч. для испытания на подлинность, испытания на чистоту и для количественного определения.
27 Оптические методы
Рефрактометрия основана на наличии зависимости величины показателя преломления света от концентрации раствора испытуемого вещества. Показатель преломления зависит также от температуры, длины волны света, концентрации вещества и природы растворителя. Рефрактометрию используют для установления подлинности лекарственных веществ по молярной рефракции. Для количественного определения выбирают интервал линейной зависимости между концентрацией раствора и коэффициентом преломления. В этом интервале концентрацию (х) вычисляют по формуле: х=(п — по)/Р, где л — показатель преломления раствора вещества; по — показатель преломления растворителя; Б — фактор, равный величине прироста показателя преломления при увеличении концентрации вещества на 1% (устанавливается экспериментально).
Рефрактометрические определения выполняют на рефрактометрах, при стабильной температуре (20±0,3°С) и длине ? злны линии Б спектра натрия (589,3 нм) в диапазоне показателей преломления от 1,3 до 1,7. Прибор юстируют по эта- - м~:ным жидкостям или воде очищенной, для которой ПО20 = 1,3330.
Поляриметрия — метод, основанный на способности вещества вращать плоскость поляризованного света. Эта способ- -: ;гь обусловлена наличием в молекулах ассиметрических атомов углерода. Степень отклонения плоскости поляризации
первоначального положения выражается в угловых градусах. Эту величину называют углом вращения (а). Правовращающие вещества вращают плоскость поляризации по часовой стрелке (обозначают знаком +), левовращающие — против часовой стрелки (-).
Для растворов величина а зависит от природы растворителя, концентрации оптически активного вещества и длины рабочего слоя кюветы с раствором. Подлинность и чистоту лекарственных веществ подтверждают по величине удельного вращения [а]о20, измеренного при 20"С и длине волны Б спектра натрия. Величину [а] о20 для растворов веществ рассчитывают по формуле:

где а — измеренный угол вращения, в градусах; / — длина рабочего слоя кюветы, в дециметрах; С — концентрация раствора вещества (г/100 мл).
Количественно определяют (в %) содержание оптически активного вещества в растворе по формуле:

Величину а измеряют на поляриметрах с точностью до ±0,02°.
28 Методы, основанные на поглощении электромагнитного излучения
Используют спектрофотометрические методы анализа по поглощению веществами монохроматического электромагнитного излучения (в УФ- и ИК-области) и фотоколориметрические (колориметрические) методы анализа по поглощению веществами немонохроматического излучения.
Фотометрические методы анализа основаны на использовании объединенного закона Бугера-Ламберта-Бера:
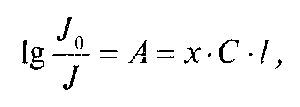
где/о — интенсивность излучения, падающего на вещество; /— интенсивность излучения, прошедшего через вещество; А — величина оптической плотности; х — показатель поглощения данного вещества; С — концентрация раствора анализируемого вещества, г; I — длина рабочего слоя кюветы, см.
На основании этого закона содержание вещества в растворе определяют по формуле:

В случае несоответствия закону Бугера-Ламберта-Бера вначале с помощью стандартного раствора устанавливают зависимость оптической плотности от концентрации, а затем строят калибровочный график, с помощью которого выполняют расчеты.
Спектрофотометрия в УФ- и видимой областях — один из наиболее широко используемых физико-химических методов в фармацевтическом анализе.
Анализируемые ЛВ должны иметь в структуре молекулы хромофорные группы (сопряженные связи, ароматическое ядро и др.), обусловливающие различные электронные переходы в молекулах и поглощение электромагнитного излучения.
Кривая зависимости интенсивности светопоглощения от длины волны (нм) называется спектром поглощения вещества и является его специфической характеристикой. Измерение спектров поглощения растворов анализируемых веществ в ультрафиолетовой (190-380 нм) и видимой (380-780 нм) областях производят с помощью спектрофотометров различных марок (СФ-26, СФ-46 и др.). В качестве растворителей используют свободные от примесей воду, растворы кислот и щелочей, этанол, хлороформ и другие органические растворители.
Спектрофотометрической константой является удельный показатель поглощения ( ), который рассчитывают по формуле:

Удельный показатель поглощения представляет собой величину оптической плотности раствора, содержащего
1,0 г вещества в 100 мл раствора, измеренную в кювете с рабочей длиной 1 см. Установив по стандартному образцу величину и преобразовав эту формулу, можно рассчитать концентрацию анализируемого вещества с относительной погрешностью до ±2%.
Идентификацию ЛВ можно провести по Д1^, характеру спектральных кривых в различных растворителях, положению максимума и минимума светопоглощения или их отношению (при различных длинах волн). Для количественного спек- трофотометрического анализа важен выбор аналитической полосы поглощения. Последняя должна быть свободна от наложения полос поглощения других компонентов смеси и иметь достаточно высокий удельный показатель поглощения анализируемого вещества.
Фотоколориметрия отличается от спектрофотометрического анализа тем, что анализируемое вещество с помощью какого-либо реагента переводят (количественно) в окрашенное соединение. Вначале получают окрашенные растворы, используя растворы стандартных образцов (ГСО или PCO). Измерение оптической плотности производят на фотоколориметрах. Затем строят калибровочный график зависимости интенсивности поглощения окрашенных растворов от концентрации, по которому рассчитывают содержание JTB в испытуемых образцах JIB или ЛФ.
Метод дифференциальной спектрофотометрии и фотоколориметрии основан на измерении светопоглощения анализируемого раствора относительно раствора сравнения, содержащего определенное количество стандартного образца испытуемого вещества или его заменителя. Такой прием приводит к изменению рабочей области шкалы прибора и снижению относительной погрешности определения до ±0,5-1%, т.е. сопоставимой с титриметрическими методами.
Производная УФ-спектрофотометрия является одним из вариантов дифференциальной спектрофотометрии. Если в дифференциальной спектрофотометрии используют разность оптических плотностей при одной и той же длине волны, то в производной — при двух длинах волн, разделенных небольшим интервалом. Этот вариант основан на выделении индивидуальных полос из УФ-спектра, который представляет собой сумму налагающихся полос поглощения или полос, не имеющих четко выраженного максимума. При этом на спектральных кривых в координатах производная-длина волны появляются полосы с отчетливо выраженными максимумами и минимумами. Благодаря этому можно идентифицировать сходные по химической структуре вещества, повысить избирательность анализа и выполнять количественное определение двух-, трехкомпонентных смесей более экономично и эффективно, чем титриметрическими методами.
Одним из вариантов дифференциальной спектрофотометрии является АЕ-метод. Он основан на превращении одного из веществ, входящих в состав анализируемой пробы, в таутомерную (или иную) форму, отличающуюся по характеру и интенсивности светопоглощения. Затем измеряют светопоглощение раствора одной таутомерной формы по отношению к другой, т.е. используют в качестве стандарта раствор анализируемого вещества.
Спектрофотометрии в ИК-области. Природа полос поглощения в ИК области связана с колебательными переходами и изменением колебательных состояний ядер, входящих в молекулу поглощающего вещества. Поэтому поглощением в ИК-области обладают молекулы, дипольные моменты которых изменяются при возбуждении колебательных движений ядер. Область применения ИК-спектроскопии аналогична, но более широка, чем у УФ-метода. ПК-спектр однозначно характеризует всю структуру молекулы, включая незначительные ее изменения. Важные преимущества ИК-спектроскопии - - высокая специфичность, объективность полученных результатов, возможность анализа веществ в кристаллическом состоянии. Для измерения ИК-спектров на однолучевых или двулучевых ИК-спектрофотометрах используют взвеси веществ в вазелиновом масле или помещают анализируемое вещество между пластинами из бромида калия. Каждый ИК-спектр представляет собой серию полос поглощения, максимумы которых определяются волновым числом, измеряемым в см"1, и определенной интенсивностью. Для анализа ЛВ обычно используют спектральную область от 4000 до 400 см-1.
ГФ XI рекомендует два способа установления подлинности по ИК-спектрам. Один из них основан на сравнении зарегистрированных в идентичных условиях ИК-спектров испытуемого ЛВ и его стандартного образца. Второй способ заключается в сравнении ИК-спекгра испытуемого ЛВ с его стандартным спектром, прилагаемым к ФС и зарегистрированным в соответствии с указанными в ней требованиями.
Фототурбидиметрия — метод, основанный на измерении интенсивности света, поглощенного тонкодисперсной суспензией, и фотонефелометрия — метод, основанный на измерении света, рассеянного взвешенными частицами анализируемого вещества. Оба метода применяют в фармацевтическом анализе для количественного определения ЛВ, образующих с различными реактивами тонкие суспензии. Предварительно устанавливают зависимость между интенсивностью поглощения (рассеяния) света и концентрацией вещества в анализируемом растворе. Способы расчета аналогичны фотометрическим методам.
30 Методы, основанные на испускании излучения
Атомно-абсорбционная спектрометрия основана на поглощении атомами излучения с частотой, равной частоте резонансного перехода. Излучение исходит от лампы с полым катодом, проходит через пламя, в котором распыляется проба, пропускается через щель монохроматора, и выделенная из спектра резонансная линия определяемого элемента измеряется фотоэлектрическим способом. Затем устанавливается зависимость между ослаблением интенсивности излучения источника света и концентрацией испытуемого вещества.
Флуоресцентные методы основаны на способности веществ флуоресцировать в УФ-свете, обусловленной либо химической структурой самих органических веществ, либо продуктов их диссоциации, сольволиза, других превращений. Способностью флуоресцировать обладают обычно органические соединения с симметричной структурой молекул, в которых имеются сопряженные связи (нитро-, нитрозо-, азо-, амидные, карбонильные или карбоксильные группы).
Флуориметрия используется не только для установления подлинности, но и определения малых количеств веществ, т.к. интенсивность флуоресценции имеет линейную зависимость от концентрации. Линейная зависимость сохраняется при постоянстве квантового выхода и интенсивности возбуждающего света для низких концентраций веществ. При высоких концентрациях эта зависимость нарушается. Идентификацию проводят по цвету излучаемого света, специфичного для флуоресцирующих веществ. Спектр дает широкие полосы излучения (от 100 до 200 нм). Метод отличается очень высокой чувствительностью. Количественное определение выполняют на спектрофлуориметрах. Расчет концентрации производят с помощью калибровочного графика или шкалы стандартных растворов, аналогично фотометрическим методам.
31 Методы, основанные на использовании магнитного поля
Спектроскопия ядерного магнитного резонанса (ЯМР) — метод, основанный на регистрации индуцированных радиочастотным полем переходов между ядерными магнитными энергетическими уровнями молекул вещества, помещенного в магнитное поле. Метод позволяет изучать магнитные переходы ядер со спиновыми квантовыми числами больше нуля (ядра 'Н, 13С, 19Р, 31Р). Совокупность сигналов переходов между энергетическими уровнями ядер молекул составляет спектр ЯМР. Каждый спектр ЯМР регистрируется для одного типа ядер и специфичен для каждого вещества. Чаще всего используют спектроскопию на протонах (ПМР) и ЯМР 13С.
Спектры регистрируют при помощи ЯМР-спектрометров. Каждый спектр является отражением числа ядер, порядка их связи и геометрии расположения ядер в молекуле. Спектр представляет собой совокупность пиков с различной шириной, площадью и интенсивностью сигналов. По характеру протонных сигналов можно сделать заключение о наличии в молекуле тех или иных групп атомов. Величина химического сдвига имеет порядок 10~6 или млн~! (миллионная доля). Она зависит от наличия в молекуле тех или иных групп, например, химический сдвиг у ароматических протонов находится в интервале 7-9 млн4, альдегидных — 9-10 млн-1 и т.д.
Метод ЯМР- и ПМР-спектроскопии используют для объективной идентификации органических Л В и для количественного определения относительного содержания вещества или примеси. Подлинность может быть подтверждена либо пут ем сравнения со стандартным образцом, либо по наиболее характерным сигналам спектра, либо по полному набору спектральных параметров.
Масс-спектроскопия — метод, позволяющий определить массу ионов, ионизированных молекул или фрагментов молекул по отклонению в магнитных и электрических полях или по кинетической энергии. Ионизация молекул происходит в результате воздействия пучка электронов. Интенсивность пика в масс-спектре пропорциональна ч ислу образовавшихся ионов данного вида. Состав и массовые числа характеристических ионов позволяют установить принадлежность исследуемого соединения к определенному классу веществ, осуществить его идентификацию. Масс-спектроскопия отличается большой информативностью и очень высокой чувствительностью.
32 Электрохимические методы
Потенциометрия — метод, основанный на измерении равновесных потенциалов, возникающих на границе между испытуемым раствором и погруженным в него электродом. В фармацевтическом анализе наиболее широко используют по- тенциометрическое титрование. Оно основано на установлении эквивалентного объема титранта путем измерения ЭДС, возникающей при титровании за счет разности потенциалов индикаторного электрода и электрода сравнения, погруженных в анализируемый раствор. Метод потенциометрии используют для определения рН (рН-метрия) и установления концентрации отдельных ионов.
Преимущества потенциометрического метода определения по сравнению с индикаторным состоят в возможности титрования окрашенных, коллоидных, мутных растворов, смеси нескольких компонентов в водных и неводных средах. Метод применим в различных видах титриметрии, основанных на реакциях нейтрализации, осаждения, окисления-восстановления. Электродом сравнения служит каломельный электрод, индикаторным — стеклянный. Измерение ЭДС между индикаторным электродом и электродом сравнения производят с помощью высокоомных потенциометров. Титр ант прибавляют равными объемами, причем вблизи точки эквивалентности по 0,1-0,05 мл. Около точки эквивалентности изменение ЭДС происходит наиболее сильно. Результаты титрования представляют либо графически, обозначая точку эквивалентности на кривой титрования, либо расчетным методом.
Ионометрия основана на использовании зависимости между ЭДС гальванической цепи с ионселективным электродом и концентрации анализируемого иона в электродной ячейке цепи. Метод отличается высокой чувствительностью, экспрессностыо, хорошей воспроизводимостью, несложным оборудованием, доступными реагентами. Широко применяют для определения ионов натрия, калия, кальция, галогенидов в многокомпонентных смесях, в т. ч. ЛФ.
Полярография — метод, основанный на измерении силы тока, возникающего на микроэлектроде, при электровосстановлении анализируемого вещества в растворе. Растворителем служит вода или органические и смешанные растворители. Электролиз проводят в полярографической ячейке, состоящей из электролизера и двух микроэлектродов: ртутного капающего и внешнего насыщенного каломельного. При соблюдении идентичных условий измерений для идентификации используют величину потенциала полуволны, а для количественного определения — высоту волны (измерение предельного диффузного тока). Количественный анализ выполняют методами калибровочных кривых с использованием стандартных растворов и методом добавок.
33 Термические методы анализа
Термические методы основаны на изменениях, которые вызывает нагревание вещества в зависимости от их природы, температуры, условий нагревания. При этом происходят полиморфные превращения, удаление сорбционной и кристаллизационной воды, сублимация, плавление, кипение, разложение. Разложение веществ сопровождается такими химиче- скичи превращениями, как структурирование, термическая, окислительная или гидролитическая деструкция. Термическая деструкция сопровождается поглощением или выделением теплоты, а также образованием газообразных продуктов. Эти процессы лежат в основе термографии — оценке термической стабильности по температурам термоэффекта, связанного с деструкцией вещества.
Термический анализ основан на точной (до 0,1°С) регистрации равновесного состояния между кристаллической и жид- кон фазами анализируемого вещества при медленном нагревании или охлаждении. Лучшей воспроизводимостью отличается дифференциальный термический анализ, основанный на регистрации изменения энергии в зависимости от температуры. Одной из модификаций этого метода является дериватография, сущность которой состоит в регистрации изменений температуры образца (термических характеристик), вызванных дегидратацией, плавлением, термической деструкцией и другими процессами, происходящими при нагревании. Особенно широкие возможности создают термические методы при исследовании стабильности ЛВ.
34 Методы разделения
В фармацевтическом анализе для разделения смесей ЛВ используют экстракцию, хроматографические методы и электрофорез.
Экстракция — метод разделения, основанный на использовании экстрагента, не смешивающегося с исходной фазой и легко отделяющегося от нее и от экстрагируемых компонентов. В зависимости от исходной фазы различают экстракцию из твердого вещества и экстракцию из раствора (жидкостную). По количеству операций экстракция может быть однократной и многократной. В фармацевтическом анализе экстракцию широко используют для разделения компонентов, входящих в состав ЛФ. Кроме того, ее сочетают с фотометрией в экстракционно-фотометрическом методе, основанном на образовании испытуемым веществом цветных продуктов реакции, способных экстрагироваться каким-либо органическим растворителем. Затем в органической фазе выполняют фотометрическое определение экстрагированного продукта.
Хроматографические методы разделения веществ основаны на их распределении между двумя фазами: подвижной и неподвижной. Подвижная фаза — жидкость или газ; неподвижная — твердое вещество или жидкость, адсорбированная на твердом носителе. Относительная скорость перемещения частиц вдоль пути разделения зависит от их взаимодействия с неподвижной фазой. Поэтому каждое вещество проходит на носителе определенный путь. Отношение пути перемещения вещества к пути перемещения растворителя есть величина постоянная, обозначаемая Rf. Она является константой для данных условий разделения и используется для идентификации ЛВ.
Хроматография на бумаге. Носителем неподвижной фазы (например, воды) служит специальная хроматографическая бумага. Распределение происходит между водой, находящейся на поверхности бумаги, и подвижной фазой, которая представляет собой систему из нескольких растворителей. Испытание выполняют согласно требованиям ГФ XI (в. 1, с. 98) или ФС (ФСП). Для подтверждения подлинности одновременно хроматографируют испытуемое вещество и стандартный образец. Если они идентичны, то пятна на хроматограммах будут1 иметь одинаковый вид и равные значения Rf. Чтобы исключить влияние на ошибку определения условий хроматографирования, пользуются более объективной константой Rs, которая представляет собой отношение величин Rf испытуемого и стандартного образцов. Хроматографию используют при испытании на чистоту. О наличии примесей судят по появлению дополнительных пятен на хромато грамме. Анализируемое вещество и примесь обычно имеют разные значения Rf.
Количественное содержание вещества можно определить непосредственно на хроматограмме, используя планиметрический, денситометрический, люминесцентный и другие методы. Используют также способы, основанные на элюирова- нии анализируемого вещества из вырезанного и измельченного участка хроматограммы с соответствующим пятном. В элюате содержание испытуемого вещества определяют фотометрическим или электрохимическим методом.
Хроматография в тонком слое сорбента (ТСХ) отличается от хроматографии на бумаге тем, что процесс хромато графирования происходит на носителе (сорбенте), нанесенном тонким слоем на инертную поверхность. Твердый сорбент может быть закрепленным или незакрепленным на этой поверхности. Сорбентом служит силикагель или оксид алюминия. Для закрепления добавляют небольшие количества крахмала или сульфата кальция. Используют также пластинки промышленного изготовления типа «Силуфол УФ-254», «Сорбфил» и др.
Преимуществами ТСХ является простота приемов и оборудования, более высокая чувствительность, чем у бумажной хроматографии, устойчивость пластинок к температурным и химическим воздействиям, значительно большие возможности процессов разделения, детектирования, элюирования, меньшая продолжительность выполнения испытания. Все это создает широкие возможности в использовании ТСХ для выполнения испытаний на подлинность, чистоту, для количественного определения ЛВ в ЛФ.
Двумерное хроматографирование отличается повторным (после высушивания) пропусканием той же или иной подвижной фазы, но в перпендикулярном по отношению к первоначальному направлении. При этом используют квадратные пластины или листы бумаги.
В фармацевтическом анализе широко применяют сочетание ТСХ с физико-химическими методами анализа. Такие комбинированные методы, как хромато-спектрофотометрия, хромато-флуориметрия, хромато-масс-спектроскопия особенно эффективны в анализе ЛРС и препаратов, содержащих большое число сопутствующих компонентов.
Газожидкостная хроматография (ГЖХ) основана на распределении компонентов смеси между газовой и жидкой или твердой фазами. Распределение происходит в результате многократных актов сорбции и десорбции анализируемых веществ, которые вводятся в поток газа-носителя, испаряются и в парообразном состоянии проходят через колонку с сорбентом. Поэтому метод ГЖХ применим для анализа летучих веществ или веществ, которые могут быть переведены в газообразное состояние. Разделенные вещества элюируются из колонки потоком газа-носителя, регистрируются детектором и фиксируются на хроматограмме в виде пиков, по которым можно идентифицировать или определять содержание каждого компонента смеси.
Газовый хроматограф включает в себя систему измерения и регулирования скорости потока газа-носителя, систему ввода пробь! испытуемого образца, газохроматографическую колонку, систему термостатирования и контроля температуры в различных узлах прибора и систему детектирования, регистрации и обработки информации, полученной на приборе.
Подлинность JIB методом ГЖХ можно подтвердить либо с помощью свидетелей, либо методом относительных удерживаний. В первом случае доказательством идентичности служит совпадение времени удерживания вещества-свидетеля и одного из компонентов смеси JIB при хроматографировании каждого в отдельности в одинаковых условиях. Во втором случае вещество-свидетель добавляют к пробе, затем анализируют по рекомендуемой методике. Рассчитывают по формуле величину относительного удерживания, которая является постоянной для JTB в конкретных условиях. Количественный анализ выполняют в тех же условиях, используя для расчетов такие параметры, как площадь или высота пиков ЛВ, Площадь пиков устанавливают на хроматограмме с помощью планиметра, интегратора или умножением высоты пика на его полуширину.
Высокоэффективная жидкостная хроматография (ВЭЖХ) отличается от ГЖХ тем, что подвижной фазой служит не газ, а жидкость, причем она проходит через колонку, наполненную сорбентом, с большой скоростью за счет значительного давления. Поэтому ВЭЖХ позволяет разделять многокомпонентные смеси на индивидуальные вещества высокой степени чистоты. ВЭЖХ отличается высокой чувствительностью (до Ю-6 г). На разделение 10-15 компонентов затрачивается 20-30 мин.
Жидкостный хроматограф включает такие узлы, как дозатор, насос высокого давления, высокоэффективную колонку, детектор с регистрирующим устройством. Колонки изготавливают из нержавеющей стали, они имеют длину 10-25 см, внутренний диаметр 0,3-0,8 см и плотно набиваются адсорбентом с размером частиц 5-10 мкм. В качестве элюента используют различные углеводороды в сочетании с этанолом. Детектором обычно служит спектрофотометр с переменной длиной волны (190-900 нм), но существуют также флуориметрические, электрохимические и другие детекторы.
Подлинность испытуемых JIB подтверждают по времени выхода каждого компонента смеси из колонки, которое будет стабильно при одинаковых условиях проведения эксперимента. Количественное содержание рассчитывается по площади пика, которая пропорциональна количеству ЛВ в пробе.
Электрофорез — метод анализа, основанный на способности заряженных частиц к перемещению в электрическом поле. Скорость перемещения ионов зависит от напряженности электрического поля, величины заряда, размера частицы, вязкости, pH среды, температуры и других факторов. Электрофоретическая подвижность — величина, характерная для испытуемого вещества. Различают абсолютную (измеряемую в сантиметрах в секунду) и относительную электрофоретичес- кую подвижность (отношение к подвижности стандартного образца). По технике выполнения и аналитическим возможностям электрофорез на бумаге и в тонких слоях сорбента сходен с ТСХ. Он позволяет разделять и идентифицировать компоненты различных смесей.
35 Биологические и микробиологические методы контроля качества лекарственных веществ
Биологическую оценку качества ЛВ проводят по их фармакологической активности или токсичности. Биологические и микробиологические методы применяют в тех случаях, когда с помощью физических, химических и физико-химичес- ких методов нельзя сделать заключение о доброкачественности ЛС. Биологические испытания проводят на животных (кошки, собаки, голуби, кролики, лягушки и др.), отдельных изолированных органах (рог матки, часть кожи) и группах клеток (форменные элементы крови, штаммы микроорганизмов и др.). Биологическую активность устанавливают, как правило, путем сравнения действия испытуемых и стандартных образцов.
Биологическая оценка сердечных гликозидов в ЛП основана на их способности вызывать в токсических дозах систолическую остановку сердца животных. Испытание проводят на лягушках, кошках и голубях, устанавливая соответствующие ЕД (единицы действия): ЛЕД, КЕД, ГЕД, которые вызывают в условиях опыта систолическую остановку сердца у животных. Активность ЛП оценивают путем сравнения с активностью стандартных образцов и рассчитывают содержание ЕД в 1г, 1 мл или в 1 таблетке испытуемого ЛС. Биологической оценке подлежат листья, трава, семена, цветки различных видов наперстянки, горицвета, ландыша, строфанта, желтушника и приготовленные из них ЛС.
Биологическую активность антибиотиков устанавливают методом, основанным на сравнительной оценке угнетения роста микроорганизмов. Наиболее широко используют рекомендованный ГФ метод диффузии в агар, заключающийся в сравнении действия определенных концентраций испытуемого и стандартного образца антибиотика на тест-микроорганизм. ЕД представляет собой меру, которой выражается биологическая активность каждого антибиотика, она соответствует определенной его массе. Для большинства антибиотиков одна ЕД соответствует 1 мкг активного вещества.
Биологическую активность инсулина устанавливают, сравнивая гипогликемическое (сахароснижающее) действие испытуемого инсулина и стандартного образца инсулина. Стандартный образец представляет собой высокоочигценный инсулин, многократно проверенная активность которого (по сравнению с активностью международного стандарта) составляет не менее 25 ЕД в 1 мг. Испытания выполняют на кроликах (ГФ XI, в. 2, с. 176).
Испытания на содержание веществ гистаминоподобного действия основаны на последовательном повторном введении животным вначале гистамина (0,1 мкг/кг), а затем испытуемого раствора. Одновременно контролируют артериальное давление. Выполняют испытания на кошках обоего пола массой не менее 2 кг под уретановым наркозом. Испытанию подвергают парентеральные ЛС (ГФ XI, в. 2, с. 185).
Испытание на пирогенность основано на способности пирогенных веществ повышать температуру тела теплокровных животных. Суть испытания состоит в измерении температуры тела кроликов после введения им в ушную вену испытуемых стерильных жидкостей. Испытания проводят на трех кроликах, масса тела которых не отличается более чем на 0,5 кг. Жидкость считают непирогенной, если сумма повышения температуры у трех кроликов не превышает 1,4°С. Если эта сумма находится в пределах 1,5-2,2°С, испытание повторяют на пяти кроликах, а если превышает 2,2°С, то жидкость считается пиро генной.
Испытание пирогенности на кроликах отличается определенными трудностями. Поэтому во многие фармакопеи мира (США, Великобритании, Китая и др.) для определения пирогенности ЛС включен так называемый Л АЛ-тест (определение бактериальных эндотоксинов). В его основе лежит способность лизата амебоцитов (клеток крови) мечехвоста специфически реагировать с эндотоксинами грамотрицательных бактерий (липосахаридами). В результате взаимодействия эндотоксина и лизата появляется помутнение прозрачной реакционной смеси или происходит образование твердого геля, что служит подтверждением присутствия эндотоксина. Сырьем для производства ЛАЛ-реагента служит кровь мечехвостов — морских животных, обитающих у берегов Северной Америки, Японии, Китая, Вьетнама.
Л АЛ-гест высокоспецифичен по отношению к эндотоксинам грамотрицательных бактерий. Его чувствительность во много раз выше, чем у фармакопейного теста на кроликах, а области применения значительно шире. ЛАЛ-тест применим в производственном (постадийном) контроле содержания эндотоксинов в инъекционных ЛФ, поскольку дает возможность получения результатов в течение 1-2 часов и одновременного испытания большого количества образцов. Кроме того, этот тест обеспечивает надежность и воспроизводимость получения результатов, сочетающихся с простотой используемой методики. Реактив для ЛАЛ-теста предсталяет собой сублимационно высушенный лизат, который готов к использованию после разведения его апирогенной водой. Учитывая преимущества ЛАЛ-теста, подготовлен проект ОФС «Определение содержания бактериальных эндотоксинов» для включения в очередное издание ГФ РФ.
Испытание на токсичность проводят на белых мышах обоего пола массой 19-21 г. Испытуемый раствор вводят в хвостовую вену пяти мышам и ведут1 наблюдение за ними в течение 48 час. Если ни одна из подопытных мышей в течение этого срока не погибнет, то ЛП считается выдержавшим испытание на токсичность. В случае гибели хотя бы одной мыши испытания повторяют по определенной схеме и делают окончательное заключение о его токсичности.
Испытаниям иа микробиологическую чистоту подвергают не стерилизуемые в процессе производства ЛП (таблетки, капсулы, гранулы, растворы, экстракты, мази и др.). Эти испытания имеют своей целью определение состава и количества имеющейся в ЛФ микрофлоры. При этом устанавливается соответствие нормам, ограничивающим микробную обсеме- ненность (контаминацию). Испытание включает количественное определение жизнеспособных бактерий и грибов, выявление некоторых видов микроорганизмов, кишечной флоры и стафилококков. Испытание выполняют в асептических условиях в соответствии с требованиями ГФ XI (в. 2, с. 193) двухслойным агаровым методом в чашках Петри.
Испытание на стерильность основано на доказательстве отсутствия в ЛС жизнеспособных микроорганизмов любого вида и является одним из важнейших показателей безопасности ЛС. Этим испытаниям подвергаются все ЛП для парентерального введения, глазные капли, мази и т.д. Для контроля стерильности применяют биогликолевую и жидкую среду Са- буро, используя метод прямого посева на питательные среды. Если ЛС обладает выраженным антимикробным действием или разлито в емкости более 100 мл, то используют метод мембранной фильтрации (ГФ, в. 2, с. 187).
36 Валидация методов анализа
Валидация — это подтверждение обоснованности выбора метода анализа для установления норм качества Л С по каждому разделу НД. Она проводится при подготовке проектов НД на новые ЛС или при последующем пересмотре НД. Ва- лидации подвергаются аналитические методы, используемые для идентификации ЛВ, установления содержания в нем различных примесей, количественного определения индивидуальных ЛВ и содержания их в ЛФ, определения вспомогательных веществ и консервантов.
Валидация метода анализа предполагает оценку его специфичности, линейной зависимости результатов испытаний, аналитической области методики, правильности, воспроизводимости результатов, предела обнаружения.
Ревалидация необходима в тех случаях, когда произошли изменения в синтезе ЛВ, в составе ЛС, в аналитической методике. Параметры аналитического метода, устанавливаемые при его валидации и ревалидации, рассчитываются в соответствии с существующими правилами статистической обработки результатов анализа.
Специфичность метода анализа обусловливает его способность достоверно установить наличие ЛВ в присутствии других компонентов (примесей, вспомогательных веществ). Оценка специфичности необходима для методов, используемых при идентификации, определении примесей и количественного содержания Л В.
Линейная зависимость аналитических сигналов от концентрации ЛВ устанавливается графически. Оценивается она на основании не менее 5 испытаний, выполненных с помощью используемой аналитической методики. Параметрами, подтверждающими линейную зависимость, являются коэффициент регрессии, угол наклона линии регрессии и остаточная сумма площадей.
Аналитическая область методики охватывает интервал между верхним и нижним пределами содержания испытуемого вещества, в котором соблюдается линейная зависимость. При этом данная методика должна обеспечивать определение с требуемыми воспроизводимостью и точностью. Аналитическая область выражается в тех же единицах,что и результаты испытаний с помощью данной методики (проценты, миллионные доли).
Правильность (точность) аналитического метода характеризует близость результатов, полученных с помощью данной методики, к истинному значению. При установлении этого параметра для количественного определения субстанций, примесей могут быть использованы стандартные образцы, другие независимые методик! ьные смеси, метод добавок.
Правильность оценивается не менее чем на трех повторностях определения для трех аналитических концентраций в пределах аналитической области.
Воспроизводимость аналитического метода отражает степень совпадений результатов отдельных испытаний при многократном использовании методики. Она устанавливается при количественном определении не менее 9 аликвот образца и выражается в результате статистической обработки по величинам стандартного отклонения, коэффициента вариации и доверительного интервала.
Межлабораторная воспроизводимость аналитического метода показывает степень воспроизводимости результатов испытаний, выполненных по разработанной методике в различных лабораториях на соответствующем оборудовании, разными аналитиками, в разное время.
Предел обнаружения — минимальное содержание анализируемого вещества, которое можно обнаружить с помощью данной методики (выражается в процентах или миллионных долях). Устанавливается для химических методов визуально. Для физико-химических методов устанавливается по минимальной концентрации испытуемого вещества, которое может быть достоверно обнаружено или рассчитывается по величине стандартного отклонения и углу наклона калибровочной кривой.
Предел количественного определения — минимальное содержание (в процентах) анализируемого вещества, которое может быть определено с достаточной точностью и воспроизводимостью. Устанавливается для любых методов визуально или расчетным путем подобно установлению предела обнаружения.
Пригодность системы — интегральная часть аналитических методик, подтверждающая надежность анализа в заданных условиях его проведения.
37 Стандартные образцы
Стандартными образцами (СО) называют вещества, с которыми сравнивают испытуемые ЛС при проведении их анализа физико-химическими или биологическими методами. Условно разделяют СО на химические и биологические, но это не исключает использования одного и того же из них и для физико-химического, и для биологического анализа. В ГФ XI, вып. 2 (с. 60) даны определения терминов «государственные стандартные образцы» (ГСО). «рабочие стандартные образцы» (PCO) и «стандартные образцы веществ-свидетелей» (СОВС). Активность, или содержание вещества (%) в ГСО принимается за 100%, если нет других указаний на этикетке. Выпуск ГСО осуществляют в соответствии с требованиями ФС, которая разрабатывается предприятием-разработчиком. В качестве PCO используют образцы серийных Л В, которые соответствуют требованиям ФС (ФСП). Расчет количественного содержания ЛВ в ЛФ проводят исходя из фактического содержания его в PCO. В качестве СОВС используют ГСО, PCO и вещества, специально изготовленные в порядке, предусмотренном ФС. Применяют СОВС для определения примесей или компонентного состава испытуемых ЛС.
Ряд особенностей имеют СО на антибиотики. Среди них имеются как однокомпонентные вещества, так и сложные, состоящие из нескольких компонентов близкой природы (аминогликозиды, полиены), каждый из которых определяет как активность и токсичность, так и физико-химические константы ЛС. При разработке такого СО его активность определяется исходя из количественного содержания в нем действующего ЛВ. Кроме того, при стандартизации антибиотиков имеется тенденция к созданию единого стандарта к группе веществ, сходных по химическому строению.
Аттестацией, хранением и реализацией СО занимается НИИКЛС. Он проводит экспертизу всех проектов ФС на ГСО, разработанных предприятиями и организациями, выпускающими эти ГСО. Только после этого они утверждаются МЗ РФ в установленном порядке.
Номенклатура ГСО отечественного производства включает около 140 наименований. Они используются при выполнении физико-химических и биологических испытаний более чем 300 ЛС (прежде всего синтетических). Еще более широко используются PCO, представляющие собой образцы серийных ЛВ. Их применение отражено в более 700 ФС для испытаний подлинности, чистоты и количественного определения.