Рекристаллизационные явления в высокомолекулярных соединениях и их роль в динамике плавления
Рекристаллизационные явления в высокомолекулярных соединениях и их роль в динамике плавления
Частично кристаллический полимер представляет собой метастабильную систему, находящуюся обычно в состоянии, далеком от положения равновесия. Такая система стремится перейти в более равновесное состояние с уменьшением свободной энергии. В условиях повышения температуры этот переход происходит в основном путем рекристаллизации. Под этим термином применительно к полимерам понимают совокупность процессов плавления и новой кристаллизации, происходящих при одной и той же температуре [1—4]. Рекристаллизация во многом определяет природу явлений, происходящих при нагревании в области плавления полимера. В частности, она приводит к появлению мультиплетности пиков плавления на термограммах ДТА и динамической калориметрии [5—9].
Количественный подход к исследованию рекристаллизационных процессов, использованный нами в работах [10—14], позволил рассмотреть, основные закономерности протекания этих явлений. Цель данной работы некоторое обобщение полученных результатов, касающееся главным образом механизма и кинетики рекристаллизации. Количественные исследования, прежде всего, показали, что сфера действия рекристаллизации шире, чем до сих пор предполагали. В определенной степени она проявляется и в медленно кристаллизующихся полимерах и при быстрых скоростях нагревания. Например, в медленно кристаллизующемся полимере — полихлоропрене рекристаллизация обнаруживается до скорости нагревания 50 град/мин. Это обусловлено рядом факторов, прежде всего значительным замедлением подъема температуры образца во время плавления, что до сих пор не учитывали.
Первый этап рекристаллизации — плавление. Этот процесс в полимерах протекает в интервале температур, и при каждой температуре плавится лишь часть кристаллической фазы. Основной причиной существования интервала плавления считается различие кристаллитов по размерам [4]. Исследование энтальпии кристаллитов методом, описанным в работах [15, 16], показало, что такую же существенную роль в плавлении играет различие кристаллитов по степени дефектности. Таким образом, при повышении температуры вначале плавятся более мелкие и более дефектные кристаллиты (или более дефектная поверхность кристаллитов), и аморфная фаза обогащается массой — «сырьем», способным к кристаллизации. Поскольку частичное плавление происходит в области температур, находящейся ниже равновесной температуры плавления, появившаяся аморфная масса вновь начинает кристаллизоваться, что составляет второй этап акта рекристаллизации.
Для заключения о механизме второго этапа исходят из
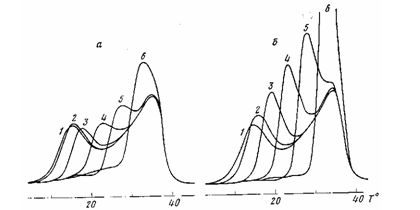
Рис. 1. Кривые плавления полихлоропрена при нагревании с V>H>= =20 град/мин. Предварительно образец был нагрет (после кристаллизации) со скоростью 10 (о) и 0,62 град/мин (б) до температур 0 (1), 5 (2), 10 (3), 15 (4), 20 (5), 25° (6). Тк>р>=0°, продолжительность кристаллизации 1 ч
предположения о хаотическом распределении кристаллитов по размерам в объеме недеформированного полимера. Результаты эксперимента [10—14] позволяют сделать вывод, что новая кристаллизация аморфной части, появившейся в результате частичного плавления, происходит путем роста наиболее мелких кристаллитов. Такую картину дает, прежде всего, статистическая обработка распределения энтальпии плавления рекристаллизовавшейся части. В сущности это наглядно видно из термограмм, получаемых при исследовании рекристаллизации методом, изложенным в работах [11, 14]. Такие термограммы, полученные на сканирующем калориметре DSC-2, приведены на рис. 1 для полихлоропренанаирит НП.
Каждая кривая получена следующим образом: образец после кристаллизации при 0° в течение 60 мин нагревали со скоростью 10 град/мин (рис. 1, а) и 0,62 град/мин (рис. 1, б) до некоторой температуры в области пика плавления, затем резко охлаждали и вторично нагревали со скоростью 20 град/мин до окончания плавления. Пик плавления, получаемый при последнем нагревании, записан на термограмме. При сопоставлении его с предыдущим пиком, полученным при нагревании до более низкой температуры, видно, что начальная часть пика исчезает из-за плавления некоторой части кристаллической фазы при предварительном более медленном нагревании. Далее, однако, пик плавления превышает предыдущий. Эта избыточная площадь соответствует рекристаллизовавшейся массе при первом нагревании.
Анализ избыточных площадей наглядно показывает, что основная температурная область плавления рекристаллизовавшейся массы в каждом эксперименте лежит вблизи верхнего предела температуры первого нагревания. Это обозначает, что энтальпии плавления расплавившихся и рекристаллизовавшихся кристаллов мало различаются. Следовательно, рекристаллизация расплавившейся массы происходит за счет роста следующих по размерам кристаллитов. При такой кристаллизации происходит максимальное снижение внутренней энергии. Кинетические препятствия, обусловленные наличием длинных молекул, естественно, приводят к некоторому отклонению от отмеченной закономерности.
Учитывая изложенный механизм, нетрудно представить процесс рекристаллизации во всем температурном интервале. По мере повышения температуры мелкие, дефектные кристаллиты и дефектная часть кристаллитов постепенно исчезают за счет роста в основном следующих по размерам кристаллитов. Процесс рекристаллизации должен вести, таким образом, к определенному выравниванию размеров кристаллитов. Известно, что нагревание сопровождается рекристаллизацией в тех случаях, когда скорости их сопоставимы. Однако необходимо обратить внимание на то, что отношение скорости рекристаллизации V>p> к скорости нагрева Vн не остается постоянным, а непрерывно изменяется с повышением температуры. Вначале, до достижения оптимальной температуры кристаллизации, при рекристаллизации оно растет, затем уменьшается. Если в начале нагревания V>Р> выше V>н>, то при достижении некоторой температуры она уменьшается до скорости нагревания. До этой температуры расплавляющаяся масса успевает рекристаллизоваться. При этом экзотермический пик кристаллизации накладывается на эндотермический пик плавления. В результате их взаимной компенсации на термограмме никакие процессы не проявляются. Такой баланс нарушается тогда, когда V>p> становится меньше V>н>. Теперь не вся расплавляющаяся фаза успевает закристаллизоваться и на термограмме появляется пик плавления. Таким образом, начало экспериментального пика плавления Т>пл> на термограмме —

Рис. 2. Зависимость начала температуры плавления закристаллизованного при 0° полихлоропрена от скорости нагревания
Рис. 3. Отношение массы рекристаллизовавшейся фазы т>р> к массе расплавившегося при нагревании полихлоропрена
Это ни что иное, как температура, при которой скорость рекристаллизации становится меньше скорости нагревания [12]. Если V>p><V>n> с самого начала нагревания, начало пика плавления будет соответствовать температуре кристаллизации. Некоторый, обычно небольшой, интервал между ними может появиться вследствие отличия номинальной температуры кристаллизации от фактической, обусловленного подъемом температуры в образце во время экзотермического процесса кристаллизации [17].
Из изложенного выше вытекает, что при нагревании полимера должны получиться термограммы двоякого типа. Если скорость кристаллизации полимера достаточно велика, так что V>p> сопоставима или больше скорости повышения температуры, то между температурой кристаллизации и пиком плавления на термограмме появится температурный интервал — предпиковая область. Она будет тем больше, чем выше скорость рекристаллизации. В случае медленно кристаллизующихся полимеров, когда V>P><V>н>, предпиковой области не будет. Своеобразную картину изменения температуры плавления в зависимости от изменения скорости повышения температуры можно получить, если скорость рекристаллизации полимера находится в диапазоне скоростей нагревания, используемых в термическом анализе. Такой случай приведен на рис. 2. При V>H>=0,31, 0,62 и 1,25 град/мин V>p> полихлоропрена оказывается выше скорости повышения температуры, и на термограмме появляется предпиковый интервал. При V>H> =2,5 град/мин, наоборот, оказывается V>р><V>н> и плавление начинается сразу после кристаллизации. Небольшой подъем T>пл>н при увеличении скорости нагревания обусловлен перегревом образца. Исследование при V>H>=0,62 град/мин дает картину (рис. 1,6), свидетельствующую о том, что рекристаллизация начинается сразу после кристаллизации, как и при скоростях нагревания, когда предпиковая область не появляется (рис. 1, а).
На рис. 3 для V>H>=0,62 град/мин сопоставлены интенсивность рекристаллизации и вид термограммы в интервале от температуры кристаллизации до полного окончания плавления. Видно, что в предпиковой области

Рис. 4. Понижение начала температуры плавления при увеличении У>н >для ПЭТФ, Г>К>р=187° {1) и полибутадиена СКД с содержанием 90% цис-1,4-звеньев, 7,>кр>=-55° (2)
Рис. 5. Зависимость формы пика плавления от температуры кристаллизации для полибутадиена СКД F„=20 град/мин. Значения Т>кр> -50 (1), -60 (2), -70 , -80 (4) и -90° (5)
интенсивность рекристаллизации вначале повышается, достигает максимума и по мере удаления от оптимальной температуры рекристаллизации начинает понижаться. При температуре 17° соотношение V>р>/V>н> уменьшается настолько, что не вся расплавляющаяся фаза успевает рекристаллизоваться, и на термограмме появляется пик плавления. При этом равновесие нарушается в пользу эндотермического процесса, что вновь приводит к увеличению вышеуказанного соотношения, следовательно, и интенсивности рекристаллизации. Эта величина, достигнув некоторого максимума, снова начинает уменьшаться по мере повышения температуры и уменьшения константы кристаллизации. Как видно из рис. 3, любые изменения интенсивности рекристаллизации приводят к определенному изменению пика плавления. Заметим, что некоторую (небольшую [14]) долю усовершенствования кристаллитов при нагревании составляет реорганизация. В данной работе она отдельно не рассматривается. Кинетика рекристаллизации обусловливается скоростью нагревания, зависимостью константы скорости кристаллизации от температуры и описанным выше механизмом процесса. Изменение аморфной части при повышении температуры dm/dT определяется частичным плавлением (dm/dT)i и рекристаллизацией (dm/dT) >2>

Определим, от каких факторов зависит начало пика плавления для полимеров с предпиковой областью. Количество рекристаллизующейся фазы пропорционально расплавившейся и способной к кристаллизации части полимера

где к — константа кристаллизации при рекристаллизации для рассматриваемой температуры, τ — время.
Примем
за начало отсчета температуру оптимальной
кристаллизации Т>кр>оп
и
будем считать .
Из эксперимента следует, что в интересующей
нас области температур m
(θ) и k(θ)
с
достаточной точностью аппроксимируются
уравнениями
.
Из эксперимента следует, что в интересующей
нас области температур m
(θ) и k(θ)
с
достаточной точностью аппроксимируются
уравнениями

где α, β, к>0>, m>0>— константы, причем к>0> и пг>0> относятся к оптимальной температуре кристаллизации, т>0> соответствует также рассматриваемой скорости нагревания.
Начало пика плавления по температуре практически совпадает с началом перегиба Т>п> на кривых ДТА и динамической калориметрии (из-за замедления подъема температуры в образце, обусловленного плавлением).

Учитывая,
что для Т>п>
имеет
место равенство окончательно
находим
окончательно
находим

где
 |
| ;
Т>а>
—
масса расплавившейся фазы при Т>п.>
;
Т>а>
—
масса расплавившейся фазы при Т>п.>
Рассмотрим, как должна изменяться кривая ДТА или динамической калориметрии после начала пика плавления. По мере замедления скорости роста температуры в образце, обусловленного эндотермическим характером процесса, количество рекристаллизующейся массы растет. Поскольку скорость роста пропорциональна массе, способной к кристаллизации аморфной фазы, а количество такого «сырья» по мере протекания процесса накапливается, то в общем случае скорость кристаллизации может равняться, а далее и превосходить скорость плавления (dm/dT)>2>>(dm/dT)>i>. Следовательно, на термограмме должен появиться максимум. В дальнейшем, однако, в связи с увеличением скорости подъема температуры в образце и уменьшением константы скорости кристаллизации скорость плавления вновь будет превосходить скорость кристаллизации. На кривой записи пика плавления должен появиться минимум, а весь пик будет иметь форму дублета. Если Т>кр> была ниже оптимальной, то в начале нагревания должен появиться еще один пик.
Температуру минимума и первого максимума можно определить на основе расчетов, аналогичных вышеприведенным, исходя из условий
Тогда



где
 —
константы скорости
—
константы скорости
кристаллизации при рекристаллизации в начале плавления Т>пл>н и при температуре первого пика Т>макс> соответственно; γ — коэффициент аппроксимации на кривой изменения скорости нагревания в образце с момента начала пика плавления;

где Т>макс>, Т>кшп> — количество расплавившейся массы при Т>ткс> и Т>мин> (температура минимума).
Приведенные формулы по заданным значениям к(Т), Г>кр>, m, V>н> позволяют рассчитать температуру начала и экстремальные точки пика и тем самым теоретически получить кривую плавления, причем видно, что с увеличением скорости нагревания Т>пл>н не увеличивается, как обычно полагают, а уменьшается. Температура первой вершины дублетного пика плавления с ростом V>н> сдвигается в область более высоких температур. Чем, больше количество расплавившейся массы, т. е. чем ниже была T>кр>, тем ближе эта вершина к началу пика. Ясно также, что в двух случаях — при низких и при высоких Т>кр> — должны получаться одинарные пики.
Таким образом, наличие одинарного пика не является показателем отсутствия рекристаллизации. Оно может быть обусловлено тем, что скорость кристаллизации при данных к, m и Vн не может стать больше скорости нагревания. На рис. 4, 5 и в таблице приведены результаты экспериментальной проверки некоторых закономерностей, вытекающих из полученных соотношений, а именно на рис. 4 проиллюстрирован эффект снижения начала температуры плавления с ростом скорости нагревания, а на рис. 5 показано изменение формы термограмм в области пика плавления в зависимости от температуры кристаллизации.
Литература
Stuart Н.A. Die Physik der Hochpolymeren. В.3. Berlin - Gottingen - Hudelberg: Springer - Verlag, 1955.
Zachmann H.G. In Fortschr. der Hochpolymeren — Forschung. Berlin: 1964, B. 3, S. 581.
Тейтельбаум Б.Я. Докл. АН СССР, 1968, т. 181, № 2, с. 368.
Годовский Ю.К. Теплофизическне методы исследования полимеров. М.: Химия, 1976 с. 95.
BairH. Salovey В.J. Polymer Sci. В, 1967, v. 5, № 5, p. 429.
Сочава И.В., Церетели Г.И., Смирнова О.И. Физика твердого тела, 1972, т. 14, вып. 2, с. 553.
Тейтельбаум Б.Я., Тузова Л.Л. Высокомолек. соед. А, 1973, т. 15, № 4, с. 917.
Сочава И.В. Физика твердого тела, 1973, т. 15, вып. 5, с. 1559.
Сочава И.В., Смирнова О.И. Физика твердого тела, 1973, т. 15. вып. 10, с. 3003.
Ягфаров М.Ш. Высокомолек. соед. А, 1975, т. 17, № 10, с. 2148.
И. Ягфаров М.Ш. В кн.: Тез. II Всес. конф. по термодинамике орган, соединений. Горький: Изд-во Горьк. ун-та, 1976, с. 94.
Ягфаров М.Ш. Высокомолек. соед. А, 1979, т. 21, № 4, с. 888.
Ягфаров М.Ш., Власов В.В., Ягфарова Т.А. В кн.: Структура и механические свойства высокомолекулярных соединений. Киев: Наукова думка, 1976, с. 18.
Ягфаров М.Ш. Высокомолек. соед. А, 1980, т. 22, № 11, с. 2609.
Ягфаров М.Ш. Высокомолек. соед. А, 1969, т. 11, № 6, с. 1195.
Ягфаров М.Ш. Высокомолек соед. А, 1979, т. 21, № 10, с. 2379.
Тейтельбаум Б.Я. Высокомолек. соед. Б, 1974, т. 16, № 10, с. 763.
1