Влияние температуры на скорость химической реакции
Влияние температуры на скорость
Правило Вант-Гоффа:
при повышении Т на
 скорость хим. реакции увеличивается в
2-4 раза. Математически это правило можно
записать:
скорость хим. реакции увеличивается в
2-4 раза. Математически это правило можно
записать:
 ,
,
 ,
,

 -
температурный коэффициент хим. реакции.
Правило Вант-Гоффа является приближённым
и его обычно используют для приблизительно
оценки скорости при изменении температуры.
Более точным является уравнение
Аррениуса, по которому:
-
температурный коэффициент хим. реакции.
Правило Вант-Гоффа является приближённым
и его обычно используют для приблизительно
оценки скорости при изменении температуры.
Более точным является уравнение
Аррениуса, по которому: .
Они могут быть вычислены по значению
констант скорости при 2-х различных Т.
При
.
Они могут быть вычислены по значению
констант скорости при 2-х различных Т.
При
 :
:
 (1). При
(1). При
 :
:
 (2). Вычитая из (1) (2) получаем
(2). Вычитая из (1) (2) получаем
 .
Отсюда можно выразить А. Зная А, по
уравнению (1) или (2) вычисляют В. Уравнение
Аррениуса может быть получено т/д-им
выводом из уравнения изобары (изохоры)
хим. реакции. Опуская индексы,
характеризующие условия протекания
реакции, это уравнение записывается:
.
Отсюда можно выразить А. Зная А, по
уравнению (1) или (2) вычисляют В. Уравнение
Аррениуса может быть получено т/д-им
выводом из уравнения изобары (изохоры)
хим. реакции. Опуская индексы,
характеризующие условия протекания
реакции, это уравнение записывается:
 ,
,
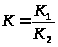 ,
где
,
где
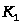 и
и
 -
константы скорости прямой и обратной
реакции. Учитывая эти уравнения можно
записать:
-
константы скорости прямой и обратной
реакции. Учитывая эти уравнения можно
записать:
 .
Представим тепловой эффект реакции Q
как разность 2-х энергетических величин:
.
Представим тепловой эффект реакции Q
как разность 2-х энергетических величин:
 .
Тогда последнее уравнение можно записать
в виде:
.
Тогда последнее уравнение можно записать
в виде:
 .
С точностью до некоторой постоянной
величины можно записать:
.
С точностью до некоторой постоянной
величины можно записать:
 ,
,
 .
Опыт показывает что
.
Опыт показывает что
 .
Отбрасывая индексы, последнее уравнение
записывается:
.
Отбрасывая индексы, последнее уравнение
записывается:
 (1), где К – константа скорости хим.
реакции. Энергетическая величина Е в
этом уравнение называется энергией
активации. Полученное уравнение описывает
зависимость К хим. реакции от температуры.
Разделив переменные и проинтегрировав,
получим:
(1), где К – константа скорости хим.
реакции. Энергетическая величина Е в
этом уравнение называется энергией
активации. Полученное уравнение описывает
зависимость К хим. реакции от температуры.
Разделив переменные и проинтегрировав,
получим:
 ,
,
 (2).
(2).
Уравнение (2) по форме
походит на уравнение Аррениуса, интегрируя
(2) получим:
 ,
,
 (3).
(3).
Уравнение используют
либо для вычисления энергии активации
по известным константам скорости при
двух температурах, либо для вычисления
константы скорости реакции при неизменной
температуре, если известна энергия
активации. Для большинства хим. реакций
энергия активации определяется в
пределах
 .
Физический смысл энергии активации
раскрывается в теории химической
кинетики, её можно определить как
некоторый избыток энергии по сравнению
со средним значением для денных условий,
которыми должны обладать молекулы чтобы
вступить в хим. реакцию. Уравнение (2)
чаще представляют в виде:
.
Физический смысл энергии активации
раскрывается в теории химической
кинетики, её можно определить как
некоторый избыток энергии по сравнению
со средним значением для денных условий,
которыми должны обладать молекулы чтобы
вступить в хим. реакцию. Уравнение (2)
чаще представляют в виде:
 .
При этом
.
При этом
 называют предэкспоненциальным множителем.
Связь энергии активации с тепловым
эффектом можно проиллюстрировать с
помощью представлению о энергетическом
барьере, который разделяет начальное
и конечное состояние системы. I и II –
уровни энергии вещ-в исходных и продуктов
реакции.
называют предэкспоненциальным множителем.
Связь энергии активации с тепловым
эффектом можно проиллюстрировать с
помощью представлению о энергетическом
барьере, который разделяет начальное
и конечное состояние системы. I и II –
уровни энергии вещ-в исходных и продуктов
реакции.
 -
энергия активации прямой реакции.
-
энергия активации прямой реакции.
 -
энергия активации обратной реакции.
Избыток энергии реагирующих молекул,
названный энергией активации, необходим
для преодоления отталкивания электронных
облаков взаимодействующих молекул при
их столкновении, и для разрыва старых
связей молекул. Уравнение Аррениуса
справедливо в области невысоких
температур; при достаточно высоких
температурах константа скорости
перестаёт зависеть от температуры.
-
энергия активации обратной реакции.
Избыток энергии реагирующих молекул,
названный энергией активации, необходим
для преодоления отталкивания электронных
облаков взаимодействующих молекул при
их столкновении, и для разрыва старых
связей молекул. Уравнение Аррениуса
справедливо в области невысоких
температур; при достаточно высоких
температурах константа скорости
перестаёт зависеть от температуры.

Теория активных столкновений
Для начала реакции
необходимо столкновение реагирующих
молекул. Но не каждое из столкновений
заканчивается хим. взаимодействием. И
хим. реакция всегда протекает со
значительно меньшей скоростью, чем это
соответствует теоретически возможному
числу встреч молекул. Например:
 при
при
 скорость
реакции составила бы примерно
скорость
реакции составила бы примерно
 ,
если учесть что каждое из столкновений
приводит к хим. превращениям. Практически
в этих условиях эта реакция протекает
со скоростью примерно
,
если учесть что каждое из столкновений
приводит к хим. превращениям. Практически
в этих условиях эта реакция протекает
со скоростью примерно
 ,
т е в
,
т е в
 раз
медленнее. Кроме того из молекулярно-кинетической
теории газов следует, что число соударений
примерно равно корню квадратному из Т,
в то время как скорость реакции растет
с Т по показанному закону. Известно, что
в некоторых случаях реагенты могут
находиться в соприкосновении без
взаимодействия между ними. Все эти факты
объясняет теория активных столкновений
Аррениуса. В основе теории лежит
представление о том. Что хим. взаимодействие
осуществляется лишь при столкновении
активных молекул, т е молекул обладающих
избытком энергии (энергии активации).
Такой хим. процесс по этой теории
представляется схемой: Нормальные
молекулы
раз
медленнее. Кроме того из молекулярно-кинетической
теории газов следует, что число соударений
примерно равно корню квадратному из Т,
в то время как скорость реакции растет
с Т по показанному закону. Известно, что
в некоторых случаях реагенты могут
находиться в соприкосновении без
взаимодействия между ними. Все эти факты
объясняет теория активных столкновений
Аррениуса. В основе теории лежит
представление о том. Что хим. взаимодействие
осуществляется лишь при столкновении
активных молекул, т е молекул обладающих
избытком энергии (энергии активации).
Такой хим. процесс по этой теории
представляется схемой: Нормальные
молекулы Активные
молекулы
Активные
молекулы Продукт
реакции. Скорость 1 процесса больше
скорости 2, и в системе всегда сохраняется
равновесие между нормальными и активными
молекулами. Следует, что для сообщения
молекулам энергии активации, её нужно
подводить извне. Известно, что в системах,
состоящих из очень большого числа
частиц, энергия распределена между ними
не равномерно. Превышающая средняя
энергия сообщается некоторой частью
молекул, в следствие перераспределения
при столкновении. Среди
Продукт
реакции. Скорость 1 процесса больше
скорости 2, и в системе всегда сохраняется
равновесие между нормальными и активными
молекулами. Следует, что для сообщения
молекулам энергии активации, её нужно
подводить извне. Известно, что в системах,
состоящих из очень большого числа
частиц, энергия распределена между ними
не равномерно. Превышающая средняя
энергия сообщается некоторой частью
молекул, в следствие перераспределения
при столкновении. Среди
 молекул при определённой температуре
встречаются
молекул при определённой температуре
встречаются
 активных молекул. По Больцману
активных молекул. По Больцману
 ,
-
число молекул, которые имеют избыток
энергии. В следствие этого скорость
реакции должна быть примерно равна
непосредственно концентрации С.
,
-
число молекул, которые имеют избыток
энергии. В следствие этого скорость
реакции должна быть примерно равна
непосредственно концентрации С.
 ,
,
 .
Тогда скорость хим. реакции
.
Тогда скорость хим. реакции
 можно
записать:
можно
записать:
 ,
,
 -
коэффициент пропорциональности.
Сравнивая эти выражения с выражением
для скорости этой реакции по закону
действия масс.
-
коэффициент пропорциональности.
Сравнивая эти выражения с выражением
для скорости этой реакции по закону
действия масс.
 ,
,
 .
Величина Е складывается из величин
.
Величина Е складывается из величин
 и
и
 ,
взятых в определённом соотношении.
Коэффициент
в теории активных соударений принимают
равным:
,
взятых в определённом соотношении.
Коэффициент
в теории активных соударений принимают
равным:
 ,
где
,
где
 -
число столкновений между реагирующими
молекулами,
-
число столкновений между реагирующими
молекулами,
 -
стерический фактор.
-
стерический фактор.
 .
.
Теория переходного состояния
В основе теории лежит закон действия масс и необх-сть активных столкновений, но сам процесс столкновения рассматривается более детально с точки зрения структурных и энерг-х уменьшений.
Рассмотрим процесс вида: А+MN=AM+N
При достаточном сближении атома А с молекулой MN,начинает ослаблять связь M-N и одновременно начинается формирование связи А-M, в результате образуется активный комплекс/переходное состояние A-M-N, в котором за счет взаимного влияния частиц и ослабления валентных связей.
Атом М в равной степени принадлежит атомам А и N, в дальнейшем наиболее вероятным течением процесса является разрыв связи M-N и образование молекулы А-М:
А+MN=A-M-N=A-M+N (1)
Мерой относительной устойчивости реакции веществ: А+MN и пер.состояние A-M-N является константой равновесия К*. От обычной константы она отличается величиной и размерностью. Само первое состояние: A-M-N также нельзя отождествлять с промежуточными хим. соединениями. Время его существования ничтожно, оно короче времени соударения, его структура составляет максимум потенциальной энергосистемы. Образование активированного комплекса в реакции требует определенной энергии активации, распад же его происходит самопроизвольно, т.е. при своем движении по пути реакции реагентная система проходит через энергетический барьер. Скорость реакции 1 равно числу активных комплексов, проходящих в единицу времени через энергетический барьер в направлении хода реакции. Она определяется как произведение так называемой линейной концентрации С* активного комплекса вдоль пути реакции.
Средняя скорость движения этого комплекса по данному пути
W=КС1С2=χС*V.
- С1 и С2 концентрация веществ: А и МN
Из последнего выражения => что константа скорости:
K=(χ(C*)V)/(C1*C2) (2) C*/(C1C2)=К* (3) К=(К*)χV (4)
V и
χ вычисляются по уравнениям
молекулярно-кинетической теории газов
и квантовой теории. К* определяется
методами статистической термодинамики
на основании спектроскопии данных.
и
χ вычисляются по уравнениям
молекулярно-кинетической теории газов
и квантовой теории. К* определяется
методами статистической термодинамики
на основании спектроскопии данных.
К= χ((RT)/(NAh))e-ΔF*/RT (5) ΔF*=ΔU*-TΔS* (6) K= χ((RT)/(NAh))e-ΔU*/RTeΔS*/R
ΔF* -изменение энергии Гельм-Гольца в процессе активации
NA –число Авогадро
h – постоянная Пианка
ΔU* -изменение внутренний энергии при активации
ΔS* -изменение энтропии при активации
Изменение внутренней энергии при активации:
ΔU=E
K=χ((RT)/(NAh))e-E/RT * eΔS*/R
Сравним полученное выражение с уравнением для константы скорости в теории активных столкновений:
K=PZ0e-E/RT
В результате сравнения получим:
PZ0=χ((RT)/(NA*h))*eΔS*/R
Отсюда следует, что стерический фактор Р действительно является вероятной характеристикой, так как определяется изменением энтропии в процессе образования активного комплекса. Возможность вычислять ΔS* и стерического фактора Р придают особую ценность теории первого состояния.
Кинетика гетерогенных р-ий
Гетерогенными называются процессы, происходящие между веществами, находящимися в различных соприкасающихся фазах. К ним относятся как хим так и физ процессы. Суммарная скорость гетерогенного процесса определяется скоростями его отдельных стадий. Если скорость одной из последующих стадий значительно меньше других, то суммарная скорость окажется скоростью этой самой медленной стадии. Если наиболее медленным звеном процесса являются стадии:
-1 или3, то кинетика суммарного процесса будет диффузионной;
-2, то скорость процесса будет определяться скоростью хим.реакции, тогда говорят, что процесс идет в кинетической области.
При сравненных скоростях диффузии и хим.превращения говорят о промежуточной области протекания гетерогенной реакции.
В случае, если гетерогенный процесс состоит из ряда одновременно протекающих параллельных процессов, то определяющим является самый быстрый процесс. Например: горение твердого и гл.топлива; восстановление твердых оксидов газами или углеродом; распределение веществ между фазами; кристаллические гл.среди хим.процессов особо нужно отметить реакции, протекающие на поверхности KAT. Отличительной особенностью гетерогенных процессов является их сложность и многостадийность.
Особенности гетерогенных процессов
Если гетерогенный процесс находится в кинетической области, то его течение подчиняется зак-м протекания х.р. на поверхности раздела фаз. Особенности таких процессов:
- любой порядок процесса
- резкая зависимость скорости процесса от температуры
γ=3

- зависимость скорости процесса от величины поверхности раздела фаз
- отсутствие влияния на скорость процесса изм-ия гидродинамических и аэродин. Условий его проведения.
Первые три особенности, хотя и в меньшей степени, наблюдаются также и тогда, когда процесс находится в переходной области. 4 особенность служит основным экспер-ым признаком того, что процесс протекает в кинетической области.
Для процессов, протекающих в диф. области отметим следующее:
1) процесс всегда
первого порядка, так как скорость
диффузии определяется 1 законом Фика:

m-масса переносимого вещества,S-площадь поверхности,
τ-время, D- коэффициент диффузии
2) слабая зависимость
скорости от температуры:
 β=1.2(в газах) β=1,5/2(в твердых телах)
β=1.2(в газах) β=1,5/2(в твердых телах)
 B-постоянная
величина; Е-значение активации диффузии
B-постоянная
величина; Е-значение активации диффузии
3) слабая зависимость скорости процесса от величины поверхности раздела фаз;
4) резкое влияние на скорость процесса аэродинамических и гидродинамических условий его проведения.
Изменение условия проведения процесса может перевести его из одной области в другую. Повышение температуры при постоянстве других факторов способствует переходу процесса в диффузионную область.
Катализ
Катализом называется
явление увеличения скорости или
возбуждения хим. р-ии происходящей под
действием некоторых веществ. Вещества,
в присутствии которых наблюдается
указанное явление называются
катализаторами. Катализаторы, участвуя
в р-ии, по её завершении вновь регенерируются,
оставаясь неизменными. Различают
гомогенный, когда катализатор и
реагирующие вещества находятся в одной
фазе, гетерогенный, когда катализатор
и реагенты в разных фазах, кроме этого
выделяют ферментативный катализ, когда
катализатором являются ферменты.
Микрогетерогенный катализ, когда
катализатор присутствует в каллойдном
состоянии, размер частиц см. особенности:
1) катализатор изменяет скорость лишь
таких р-ии, которые т/д возможны при
данных условиях
 .
Прежде чем подбирать катализатор для
той или иной р-ии нужно убедиться в её
т/д возможности.
.
Прежде чем подбирать катализатор для
той или иной р-ии нужно убедиться в её
т/д возможности.
2 )
Катализатор непосредственно участвует
в р-ях, образовывая промежуточные
соединения, изменяя тем самым число и
вид элементарных стадий процесса.
Схематично можно представить так. Пусть
для р-ии А+В=С+D катализатор является
вещество К, тогда в присутствии
катализатора схема может быть такой
А+К→АК (а), АК+В=C+D+K (б). Р-ии (а) и (б) идут
во много раз быстрее, чем р-ии
непосредственного взаимодействия без
катализатора. Промежуточное соединение
в катализе это не обычное устойчивое
хим. соединение, которое может быть
выделено в чистом виде или может
существовать в виде отдельной фазы. В
гомогенном катализе это очень не стойкие
соединения с малым временем жизни. В
гетерогенном катализе это поверхностное
соединение, не существующее в виде
отдельной фазы, свойства которого резко
отличаются от свойств аналогичного
соединения образовывающего объёмную
фазу.
)
Катализатор непосредственно участвует
в р-ях, образовывая промежуточные
соединения, изменяя тем самым число и
вид элементарных стадий процесса.
Схематично можно представить так. Пусть
для р-ии А+В=С+D катализатор является
вещество К, тогда в присутствии
катализатора схема может быть такой
А+К→АК (а), АК+В=C+D+K (б). Р-ии (а) и (б) идут
во много раз быстрее, чем р-ии
непосредственного взаимодействия без
катализатора. Промежуточное соединение
в катализе это не обычное устойчивое
хим. соединение, которое может быть
выделено в чистом виде или может
существовать в виде отдельной фазы. В
гомогенном катализе это очень не стойкие
соединения с малым временем жизни. В
гетерогенном катализе это поверхностное
соединение, не существующее в виде
отдельной фазы, свойства которого резко
отличаются от свойств аналогичного
соединения образовывающего объёмную
фазу.
3) Катализатор не изменяет величины теплового эффекта р-ии, в противном случае имело бы место не соблюдения з. сохранения и превращения энергии.
4) Катализатор не изменяет величины константы равновесия ∆H,∆S, ∆U, ∆F. Это означает, что равновесие, выход в присутствии катализатора остаётся тем же самым. Катализатор изменяет кинетические характеристики р-ии (EАКТ и предэкспоненты PZ0). действие катализатора может быть объяснено энергией активации процесса. Е3 и Е4 – энергии активации при образовании промежуточных соединений (а) и при его распаде (б), вследствие того, что энергия активации Е1 заменяет меньшую энергию Е3 и Е4 2-ух последних стадий скорость р-ии возрастает, даже если Е3+ Е4.>Е1 CH3CHO→CH4+CO при t=518ºC ЕАКТ=45.5 ккал/моль в присутствии паров J2 энергия активации снижается до ЕАКТ=32,5 ккал/моль. Скорость возрастает в 10000 раз. Катализатор ускоряет и прямую и обратную р-ии в равной мере, при этом константа равновесия не изменяется. А время достижения системой равновесного состояния уменьшается.
5) Катализатор действует избирательно. Различные катализаторы могут или одну р-ию или группу р-ий или же р-ии различного класса в соответствии с этим катализаторы могут обладать индивидуальной специфичностью, групповой специфики или являться универсальными. Особое значение катализаторов имеет и используется при протекании в параллельных р-иях.
6) В гетерогенном катализе большое влияние на процесс имеет адсорбционная способность твердых катализатора, состояние его поверхности, способов и методов обработки поверхности, присутствие на поверхности атомов других элементов и т.д.
Например: введённые в катализатор некоторые добавки, которые сами не обладают каталитической активностью, могут сильно повысить активность катализатора, такие добавки называются промоторами. Наоборот присутствие некоторых др. веществ на поверхности катализатора может сильно снизить его каталитическую активность, такие вещества называются каталитическими ядрами.