Физическая связь
Московский Государственный Технологический Университет "СТАНКИН"
Реферат по химии
"Физическая связь"
Выполнил: Фридлянд Д.А.
Проверил: Козлов Г.В.
Содержание
|
Введение |
3 |
|
§1 Ван-дер-ваальсовые взаимодействия |
4 |
|
Уравнение Ван-дер-Ваальса |
4 |
|
Ориентационные взаимодействия Ван-дер-Ваальса (эффект Кезома) |
6 |
|
Индукционные взаимодействия Ван-дер-Ваальса (эффект Дебая) |
8 |
|
Дисперсионные взаимодействия Ван-дер-Ваальса (эффект Лондона) |
9 |
|
Ван-дер-ваальсово отталкивание (эффект Паули) |
12 |
|
§2 Водородные связи |
13 |
|
Выводы |
17 |
|
Литература |
19 |
Введение
Нижеследующий реферат будет посвящен различным видам физической связи (ван-дер-ваальсовой и водородной) и их связи с физическими и химическими свойствами веществ. В отличие от химической связи (ковалентная, донорно-акцепторная, ионная) водородная и ван-дер-ваальсовая связи, как правило, не на столько сильны. Однако они оказывают значительное влияние на многие физические свойства веществ (теплота испарения жидкости либо теплота возгонки кристалла, температуры плавления и кипения). А также на количественные характеристики некоторых химических реакций: такие как тепловой эффект и энергия активации (температура активации либо минимальная для активизации реакции частота излучения) образования и диссоциации молекулярных комплексов, молекул и сложных ионов. Именно ван-дер-ваальсовые и водородные взаимодействия являются причиной как коагуляции коллоидных растворов так и их устойчивости, а также физической основой абсорбции и адсорбции, что уже сейчас применяется при проектировании очистных сооружений.
Молекулы, валентно насыщенные в обычном понимании (такие как CO>2>, H>2>O, I>2, >Ne и др.), взаимодействуют между собой, о чем свидетельствует конденсация реальных газов (идеальный газ не конденсируется ни при каких условиях). Силы, действующие между молекулами газа и вызывающие отклонение газов от идеальности, называют силами Ван-дер-Ваальса, по имени ученого, который впервые учел взаимное притяжение и отталкивание молекул при выводе уравнения состояния реальных газов.
Как известно, все химические вещества состоят из молекул в свою очередь состоящих из электрически заряженных частиц, электронов и атомных ядер. Вследствие того, что в молекуле положительные и отрицательные заряды разделены и постоянно находятся в относительном движении, в каждый момент времени подавляющее большинство молекул находятся в виде электрических диполей. Взаимодействие возникших вследствие тех или иных причин дипольных моментов молекул и называется связью Ван-дер-Ваальса. В зависимости от происхождения дипольного момента взаимодействующих молекул ван-дер-ваальсового притяжения разделяют на ориентационные, индукционные и дисперсионные. Притяжение между молекулами принимает существенные значения уже на довольно больших расстояниях (~10Å); однако же, взаимодействие Ван-дер-Ваальса включает эффекты не только притяжения, но и отталкивания, эти силы играют в нашей жизни не менее важную роль т.к. не позволяют всем молекулам слипнуться в единый материальный ком, в гигантскую глобулу.
Т о,
что плотности жидкостей и кристаллов
имеют вполне конечную величину, указывает
на одновременное существование
отталкивания между молекулами; не будь
отталкивания, молекулы при сближении
сливались бы в одно целое и плотность
возрастала бы практически не ограниченно.
В конденсированном состоянии (жидкости
или кристалле), построенном из молекул,
притяжение сближает частицы до расстояния,
на котором силы притяжения и отталкивания
равны по величине. Потенциальная
кривая взаимодействия двух молекул
приведена на рис.1; от
потенциальной кривой двухатомной
молекулы она отличается лишь количественно:
глубина потенциальной ямы, т.е. энергия
взаимодействия, значительно
меньше, а равновесное расстояние
s>0>- больше. Таким
образом, различие между химическими и
межмолекулярными (ван-дер-ваальсовыми
и водородными) связями в первую очередь
- количественное. Природа же сил в обоих
случаях - одна и та же - электрическая.
Расстояние между молекулами в жидкостях
и кристаллах ~3-5Å,
а энергия взаимодействия ~1-5
кДж/моль для сил Ван-дер-Ваальса,
что в 100 раз меньше энергии химической
связи (водородные связи как по энергии,
так и по длинне связи примыкают как к
ван-дер-ваальсовым, так и к химическим
связям практически вплотную). Для
определения энергии межмолекулярной
связи определяют
энергию сублимации (либо испарения)
и вычитают из нее работу изобарного
расширения; =-p(V>г>-V>ж>)-pV>г>-RT.
о,
что плотности жидкостей и кристаллов
имеют вполне конечную величину, указывает
на одновременное существование
отталкивания между молекулами; не будь
отталкивания, молекулы при сближении
сливались бы в одно целое и плотность
возрастала бы практически не ограниченно.
В конденсированном состоянии (жидкости
или кристалле), построенном из молекул,
притяжение сближает частицы до расстояния,
на котором силы притяжения и отталкивания
равны по величине. Потенциальная
кривая взаимодействия двух молекул
приведена на рис.1; от
потенциальной кривой двухатомной
молекулы она отличается лишь количественно:
глубина потенциальной ямы, т.е. энергия
взаимодействия, значительно
меньше, а равновесное расстояние
s>0>- больше. Таким
образом, различие между химическими и
межмолекулярными (ван-дер-ваальсовыми
и водородными) связями в первую очередь
- количественное. Природа же сил в обоих
случаях - одна и та же - электрическая.
Расстояние между молекулами в жидкостях
и кристаллах ~3-5Å,
а энергия взаимодействия ~1-5
кДж/моль для сил Ван-дер-Ваальса,
что в 100 раз меньше энергии химической
связи (водородные связи как по энергии,
так и по длинне связи примыкают как к
ван-дер-ваальсовым, так и к химическим
связям практически вплотную). Для
определения энергии межмолекулярной
связи определяют
энергию сублимации (либо испарения)
и вычитают из нее работу изобарного
расширения; =-p(V>г>-V>ж>)-pV>г>-RT.
То, что потенциальная кривая взаимодействия двух молекул не отличается качественно от потенциальной кривой двухатомной молекулы, указывает на нестрогость (размытость) границы между химическим и межмолекулярным взаимодействием; межмолекулярная связь имеет характер возмущения электронного облака одной молекулы электронным облаком другой молекулы, как ковалентная связь имеет характер возмущения электронного облака одного радикала (валентно ненасыщенная группа атомов) или атома электронным облаком другого радикала или атома. При образовании физической (межмолекулярной) связи энергия системы понижается на величину энергии возмущения, называемую энергией межмолекулярного взаимодействия; аналогично тому, как энергия системы понижается на величину энергии возмущения, называемую тепловым эффектом реакции, при образовании химической (ковалентная или донорно-акцепторная) связи. Именно на разрыв физических связей тратится теплота парообразования или сублимации, совершение работы расширения системы занимает в теплоте парообразования ничтожно малую долю.
Однако между физическими и химическими связями всеже есть одно принципиальное отличие. Это принципиальное отличие - насыщаемость химических связей в противовес абсолютной ненасыщаемости ван-дер-ваальсовых и ионных (по этому их часто тоже относят к физическим связям) взаимодействий. Водородные связи в отличие от ван-дер-ваальсовых (и ионных) взаимодействуют не со всеми молекулами конденсированной фазы (кристалл, расплав), а лишь с ближайшими соседями; однако в отличие от химических связей число ближайших соседей ограничено нестрого и может слегка варьироваться в пределах одной фазы, поэтому как правило водородные связи считают ненасыщаемыми и относят к физическим, а не к химическим связям. Однако водородные связи все таки обладают насыщаемостью (хотя и нечетко выраженной); поэтому в медицинской литературе при рассмотрении химии высоко организованных органических молекул и молекулярных комплексов (учитывая, что водородные связи примыкают к химическим также по значениям длинны и энергии связи) часто рассматривают наравне с химическими связями.
§1 Ван-дер-ваальсовые взаимодействия
Уравнение Ван-дер-Ваальса
В 1873 г. Ван-дер-Ваальс на основе молекулярной
модели несжимаемых шаров диаметра D,
притягивающих друг друга и притягиваемых
друг другом, вывел свое удивительно
простое уравнение. В реальном газе в
результате молекулярного притяжения
увеличивается кинетическое давление
по сравнению с давлением в идеальном
газе. Из самых общих соображений
молекулярное притяжение пропорционально
числу как притягивающих, так и притягиваемых
молекул; Δp ~
N2. В
результате молекулярного отталкивания
свободный объем в реальном газе меньше,
чем объем сосуда занимаемого газом.
Запрещенный объем вокруг каждой молекулы,
в который не может попасть центр другой
молекулы из-за взаимного отталкивания,
Ван-дер-Ваальс оценил как объем сферы
 , где
D - расстояние между
центрами двух несжимаемых шаров диаметра
D. Следовательно, полный
запрещенный объем моля газа будет равен
, где
D - расстояние между
центрами двух несжимаемых шаров диаметра
D. Следовательно, полный
запрещенный объем моля газа будет равен
 ,
т.е. равен учетверенному объему N>a>
несжимаемых молекул.
,
т.е. равен учетверенному объему N>a>
несжимаемых молекул.
Уравнение Клапейрона для идеального газа - pV=N>a>kT.
Уравнение Ван-дер-Ваальса представляет собой уравнение Клапейрона, в которое введены перечисленные выше поправки на возросшее вследствие межмолекулярного взаимодействия кинетическое давление и уменьшенный реальный свободный объем:
 или
или  ,
,
где a – постоянная, b4V>0> (V>0> – объем молекулы).
Если в качестве переменных P, V и T использовать их относительные значения P>c>=P/P>k >, T>c>=T/T>k> , V>c>=V/V>k> (где P>k>, T>k>, V>k> – критические значения), то закон Ван-дер-Ваальса принимает вид универсального закона соответственных состояний:
P>c>=F(V>c>,
T>c>) — универсальная
функция,  — универсальная
постоянная.
— универсальная
постоянная.
Следствие из этого закона может быть сформулировано следующим образом: все вещества кипят при одних и тех же относительных давлениях и температурах. Или еще: относительные объемы всех веществ одинаковы при одних и тех же относительных давлениях и температурах. Уравнение Ван-дер-Ваальса можно записать и в другом виде:
 ,
,
т.е. представить в виде разложения потенциала притяжения по обратным степеням температуры, в котором учтен только первый член. Оправданием такого приближения служит предположение Ван-дер-Ваальса о дальнодействующем характере сил притяжения. В случае дальнодействия можно считать, что при переходе от одной конфигурации молекул к другой их потенциальная энергия не изменится, т.е. a=const вследствие того, что они находятся в среднем поле соседей с постоянной плотностью энергии.
Математическое и экспериментальное исследования этого уравнения показали, что поправки Ван-дер-Ваальса обладают глубоким физическим смыслом. Они качественно описывают не только изменения свойств системы, определяющих фазовый переход газ-жидкость, но и форму критической области. Кроме того, если силы притяжения нельзя рассматривать постоянными из-за близкодействия, то уравнение Ван-дер-Ваальса допускает следующее приближение с учетом члена 1/T2.
Ван-дер-Ваальс в 1873 году одним из первых указал на наличие нехимического межмолекулярного взаимодействия в аморфных состояниях вещества и разделил это взаимодействие на дальнодействующее притяжение и близкодействующее отталкивание. При этом, он предложил до сих пор самую простую, но в тоже время достаточно точную в широком интервале температур и давлений, математическую модель для учета вышеперечисленных сил при расчете состояний реального газа. В связи с вышеуказанными обстоятельствами дальнодействующие силы межмолекулярного притяжения и близкодействующие силы межмолекулярного отталкивания назвали силами Ван-дер-Ваальса.
В табл.1 приведены величины -RT при температурах конденсации некоторых веществ.
|
Вещество |
Ar |
Kr |
Xe |
CH>4> |
C>2>H>6> |
C>3>H>8> |
C>5>H>12> |
H>2>O |
C>2>H>5>OH |
|
Температура конденсации, K |
87,25 |
119,75 |
165,05 |
111,57 |
184,52 |
231,09 |
309,22 |
373,15 |
351,52 |
|
-RT, Дж/моль |
6883 |
8033 |
14600 |
7268 |
13090 |
16860 |
23220 |
37560 |
35660 |
Несложно заметить, что энергия межмолекулярного взаимодействия у веществ в конденсированном состоянии сильно меняется от вещества к веществу, во многие разы, но в то же время не в порядки; этот факт указывает на то, что в разных веществах реализуются принципиально различные механизмы реализации сил межмолекулярного взаимодействия, имеющие, однако, общую природу.
Как показывает квантовомеханический расчет энергия дальнодействующего межмолекулярного взаимодействия состоит из так называемой электростатической, и энергии возмущения второго порядка - индукционной и дисперсионной. Электростатическое взаимодействие возникает между дипольными моментами молекул, между ионами и диполями в растворах или сплавах. Для нейтральных молекул в электростатическом взаимодействии важно так называемое ориентационное взаимодействие постоянных дипольных моментов молекул.
Ориентационное, индукционное и дисперсионное взаимодействия - три важнейшие составляющие ван-дер-ваальсовых сил притяжения. Силы Ван-дер-Ваальса называют дальнодействующими, так как энергия ван-дер-ваальсового взаимодействия довольно медленно спадает с расстоянием и пропорциональны r-n, для различных видов ван-дер-ваальсового притяжения n6.
Ориентационные взаимодействия Ван-дер-Ваальса
(эффект Кезома)
Р ассмотрим
возможные силы взаимодействия между
двумя диполями…
ассмотрим
возможные силы взаимодействия между
двумя диполями…
Если два диполя расположены на одной прямой и одинаково ориентированы (см. рис. 2а), то они притягиваются с силой обратно пропорциональной третьей степени расстояния между ними, установка диполей в "хвост". Аналогичная сила действует между двумя противоположно направленными диполями, расположенными на параллельных прямых, на кратчайшем расстоянии друг от друга (см. рис. 2б), установка диполей "один под другим" (антипараллельная установка диполей). В обоих случаях они ориентируются так, чтобы энергия системы стала минимальной (см. рис. 2). Если диполи ориентированы не так, как показано на рис. 2 то между диполями кроме силы радиального взаимодействия (притяжение либо отталкивание) возникает крутящий момент.
Пусть расстояние между центрами диполей (s) намного больше длинны диполя (l). Заряд разнесенный в диполе на расстояние l обозначим через e. Тогда энергию ориентационного взаимодействия можно представить как алгебраическую сумму кулоновского притяжения и отталкивания зарядов полюсов диполей:
|
U>ор> |
= |
|
e2 |
|
e2 |
+ |
2e2 |
= |
|
2e2l2 |
. |
|
s-l |
s+l |
s |
(s2-l2)s |
Пренебрегая величиной l2 по сравнению с s2 в знаменателе и обозначая через дипольный момент (=el), получаем: U>op-->= - 22/s3. Для двух разных полярных молекул с моментами >1> и >2> такой же расчет дает U>op>= 2>1>>2>/s3. При установке диполей один под другим (см. рис.2)
|
U>ор> |
= |
2 |
e2 |
+ 2 |
e2 |
= |
-2e2(s2+l2)+ 2e2s(s2+l2)1/2 |
. |
|
s |
(s2+l2)1/2 |
(s2+l2)s |
Учитывая, что l2<<s2 , воспользуемся приближенным равенством (s2+l2)1/2 s+l2/2s . Тогда
|
U>op> |
= – |
e2l2 |
, откуда следует: U>op>>>=-2/s3. Выше упомянутые формулы справедливы для расчета |
|
(s2+l2)s |
Энергии ориентационного взаимодействия тогда, когда тепловое движение не расстраивает ориентацию молекул, т.е. когда U>op>>>kT. Поэтому они пригодны для расчета энергии лишь в молекулярных кристаллах, где положение молекул фиксировано. В жидкостях и газах тепловое движение приводит ко всевозможным ориентациям молекул. При усреднении энергии взаимодействия по всем возможным ориентациям с учетом теплового движения, для жидкостей и газов получаем следующую формулу:

Для полярных молекул вклад ориентационного взаимодействия в энергию межмолекулярного взаимодействия жидкостей можно оценить, не зная конкретных расстояний между молекулами. В полярном диэлектрике на молекулу действует эффективное поле, создаваемое всеми остальными молекулами E>эфф>. Понижение энергии одного моля частиц при этом W=-½N>A>E>эфф>.(Множитель ½ введен чтобы не учитывать взаимодействие одной пары частиц дважды.) Эффективное поле можно выразить через диэлектрическую проницаемость и показатель преломления среды n>D>, в результате чего получим:
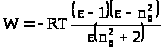
Вычисленные по этой, очень приближенной формуле значения W показывают, что ориентационный вклад в энергию сцепления полярных жидкостей весьма существенен, но все же как правило меньше вклада дисперсионных сил. Это объясняется тем, что по сравнению с общим случаем электростатических взаимодействий (например ионное взаимодействие) аддитивностью ориентационных взаимодействий можно пренебречь, т.к. взаимная ориентация двух диполей мешает им ориентироваться относительно третьего. Поэтому эффект ориентационного взаимодействия уступает действию значительно более аддитивных вследствие гибкости дисперсионных связей дисперсионных взаимодействий (речь о дисперсионных взаимодействиях пойдет ниже). Вклад ориентационного взаимодействия в энергию сцепления полярных жидкостей является преобладающим лишь для наиболее полярных молекул, например для воды.
Ориентационные взаимодействия Ван-дер-Ваальса играют определяющую роль в процессах электролитической диссоциации. Для наиболее полярных веществ связи Ван-дер-Ваальса ориентационной природы вносят наиболее существенный вклад в значения энергии и температуры плавления и сублимации (или кипения). Ориентационные взаимодействия Ван-дер-Ваальса, наряду с ван-дер-ваальсовыми индукционными взаимодействиями используются в абсорбционных и адсорбционных пылеочистных сооружениях (поэтому в качестве абсорбатов и адсотбатов в пылеочистных сооружениях практически всегда используются именно сильнополярные материалы.
Индукционные взаимодействия Ван-дер-Ваальса
(эффект Дебая)
Молекула, обладающая постоянным дипольным моментом, наводит в другой молекуле, неполярной или полярной, так называемый индуцированный дипольный момент. Величина индуцированного электрическим полем напряженности E дипольного момента >инд> может быть представлена следующим рядом: >инд> = E+E2+ . Для электрических полей малой напряженности можно пренебречь всеми членами ряда, кроме первого, это приближение можно сделать для пары индукционно взаимодействующих диполей. >инд> = E, где - поляризуемость молекулы. Индуцированный дипольный момент имеет то же направление, что и линии напряженности электрического поля постоянного диполя вызвавшего появление наведенного дипольного момента у поляризуемой молекулы или радикала в точке нахождения поляризуемой молекулы (или соответственно радикала). Взаимодействие постоянного диполя одной молекулы (радикала, сложного иона) и наведенного им диполя второй молекулы (или вообще группы атомов) понижает потенциальную энергию системы из двух молекул и упрочняет систему.
Пусть в неполярной молекуле, отстоящей
от полярной молекулы на расстоянии s,
индуцируется момент >инд>.
Энергия U>инд>
взаимодействия молекул зависит от
напряженности E поля,
создаваемого в центре неполярной
частицы постоянным
дипольным моментом полярной молекулы
и от величины индуцированного момента
>инд>:  .
Интеграл берется от 0 до E,
так как изначально поля в центре
неполярной молекулы нет, и, возникая
под внешним влиянием полярной молекулы,
оно растет от 0 до E. Подставив
в выражение для U>инд>
ранее полученное выражение
для μ>инд>, получим: U>инд>= ½E2.
Напряженность E поля
создаваемого в центре неполярной
молекулы положительным и отрицательным
полюсами постоянного диполя может быть
выражена следующей формулой:
.
Интеграл берется от 0 до E,
так как изначально поля в центре
неполярной молекулы нет, и, возникая
под внешним влиянием полярной молекулы,
оно растет от 0 до E. Подставив
в выражение для U>инд>
ранее полученное выражение
для μ>инд>, получим: U>инд>= ½E2.
Напряженность E поля
создаваемого в центре неполярной
молекулы положительным и отрицательным
полюсами постоянного диполя может быть
выражена следующей формулой:
 .
.
Здесь, как и раньше, пренебрегая
l2
по сравнению с
s2,
получим:
 .
Подставив полученное для напряженности
выражение в формулу энергии индукционного
взаимодействия (U>инд>= ½E2)
получаем:
U>инд>= 22/s6.
.
Подставив полученное для напряженности
выражение в формулу энергии индукционного
взаимодействия (U>инд>= ½E2)
получаем:
U>инд>= 22/s6.
Если учесть, что наводящая индуцированный дипольный момент молекула сама обладает поляризуемостью, формула примет окончательный вид:

Энергия индукционного взаимодействия, как и ориентационного, убывает пропорционально шестой степени расстояния, но индукционное взаимодействие не зависит от температуры. Последнее связано с тем, что ориентация наведенного дипольного момента не может быть произвольной, она однозначно определяется направлением и положением наводящего диполя в пространстве.
Величина энергии индукционного взаимодействия U>инд> тем значительнее, чем выше поляризуемость поляризуемых молекул. Индукционное взаимодействие наблюдается: при образовании гидратов благородных газов, в растворах полярных веществ, в неполярных растворителях (например, ацетона в тэтрахлорметане) и т.п., но существенно только для молекул со значительной поляризуемостью; к ним, в первую очередь, относятся молекулы с сопряженными связями.
Индукционное взаимодействие не аддитивно. Это становится ясным, если рассмотреть неполярную частицу в поле двух симметрично расположенных диполей. Каждый из них, действуя сам, вызвал бы индукционный эффект, но совместное их действие взаимно уравновешивается, в результате чего дипольный момент у неполярной частицы не наводится, а следовательно энергия системы в рассматриваемом случае индукционным взаимодействием не понижается.
В следствие нераспространенности легко поляризуемых молекул и неаддитивности индукционных взаимодействий эффект Дебая никогда не бывает доминирующим по сравнению с эффектом Кезома (ориентационные взаимодействия) и с эффектом Лондона (см. ниже).
Дисперсионные взаимодействия Ван-дер-Ваальса
(эффект Лондона)
Существуют, однако, такие молекулы у которых нет не только дипольного электрического момента, но и электрических моментов более высокого порядка; это - сферически симметричные молекулы, прежде всего молекулы идеальных газов. Однако и благородные газы при охлаждении сжижаются, а при дальнейшем охлаждении (гелий - только под повышенным по сравнению с атмосферным давлением) кристаллизуются. Силы, приводящие к конденсации идеальных газов, называются дисперсионными ван-дер-ваальсовыми силами. Дисперсионные взаимодействия Ван-дер-Ваальса играют большую роль при взаимодействии и между всеми другими молекулами, без исключений.
Решение уравнения Шредингера для системы из двух молекул методом возмущений указывает на существование электростатического, индукционного и дисперсионного взаимодействий. Каждый из указанных эффектов имеет строгое квантово-механическое определение, но если ориентационный и индукционный эффекты можно понять также на основе представлений электростатики, то дисперсионное взаимодействие объяснимо только на основе квантовой механики.
Грубое модельное представление о дисперсионном взаимодействии между двумя атомами благородного газа можно составить, рассматривая протоны ядра атома и движущиеся вокруг ядра электроны как положительный и отрицательный полюсы вращающихся вокруг центра атома мгновенных электрических диполей.
Поскольку направление этих диполей меняется с частотой 1015 циклов в секунду, вследствие чего атом не обладает дипольным моментом постоянного направления, в среднем по времени его дипольный момент равен нулю. Однако, при встрече двух атомов их мгновенные дипольные моменты ориентируются друг относительно друга, и их направления изменяются "в такт". Эта корреляция между направлениями мгновенных дипольных моментов атомов (или обладающих электронами ионов) уменьшает потенциальную энергию системы на величину, называемую энергией дисперсионного взаимодействия (или величиной эффекта Лондона).
Сущность эффекта Лондона заключается
в том, что электроны в атомах и молекулах
можно уподобить колеблющимся около
ядра частицам — осцилляторам. Любой
осциллятор, согласно современным
квантовомеханическим представлениям,
даже при абсолютном нуле температуры
совершает так называемые нулевые
колебания с энергией =h>0>/2 , где
>0>
- частота колебаний осциллятора.
При сближении двух осцилляторов и их
ориентации в такт происходит нечто
подобное соединению двух маятников
упругой нитью: из двух колебаний
осцилляторов с частотами >0>
возникают два близких к ним с
частотами >1>>>0> и >2><>0>.
Если до сближения сумма энергии
нулевых колебаний двух частиц была
равна
 ,
то, как показывает расчет, при сближении,
когда происходит взаимное возмущение
электронных облаков двух молекул,
суммарная энергия нулевых колебаний
станет равной
,
то, как показывает расчет, при сближении,
когда происходит взаимное возмущение
электронных облаков двух молекул,
суммарная энергия нулевых колебаний
станет равной  . Таким
образом, произойдет понижение энергии
нулевых колебаний системы, равное по
величине:
. Таким
образом, произойдет понижение энергии
нулевых колебаний системы, равное по
величине:  .
.
Если считать электроны в атомах и молекулах не линейными, а пространственными осцилляторами, то получим следующее выражение для энергии дисперсионного взаимодействия:
 .
.
Дисперсионное взаимодействие имеет
чисто квантовомеханическую природу —
понижение суммарной энергии нулевых
колебаний осцилляторов. Объединяя в
полученной формуле все постоянные для
данной молекулы величины в одну, получим:
U>дисп>=-C/s6,
где C -
константа. Лондон,
разработавший теорию дисперсионного
взаимодействия, показал, что величину
энергии нулевых колебаний осциллятора
можно заменить потенциалом ионизации
I. Тогда для одинаковых
частиц константа Лондона равна может
быть представлена в следующем виде:  . Для
двух неодинаковых частиц константа
Лондона представлена следующей формулой:
. Для
двух неодинаковых частиц константа
Лондона представлена следующей формулой:
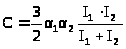 .
.
Так как потенциалы ионизации молекул колеблются в нешироких пределах, вблизи 10 эВ, то различие энергии дисперсионного взаимодействия определяется главным образом поляризуемостью молекул.
Энергия дисперсионного взаимодействия, так же как ориентационного и индукционного взаимодействий, пары частиц обратно пропорциональна шестой степени расстояния; однако же для приближенного расчета теплоты испарения жидкости следует ввести поправочный коэффициент, учитывающий координационное число и другие факторы, т.е. параметры взаимодействия частицы с ее окружением.
Особенностью дисперсионного взаимодействия является его всеобщность - во всех молекулах есть движущиеся электроны, поэтому дисперсионное взаимодействие существенно для всех без исключения молекул, а для неполярных молекул эффект Лондона - главный и практически единственный источник сил Ван-дер-Ваальса (если расплав или кристалл неполярного вещества - недостаточно очищен от полярных примесей, то индукционное взаимодействие там может тоже быть представлено, но его вклад в этом случае - пренебрежимо мал). Дисперсионное взаимодействие вносит также определенный вклад в энергию связи ионов в молекулах и в ионных кристаллах.
Дисперсионные взаимодействия играют основную роль в межмолекулярных взаимодействиях подавляющего большинства веществ. Они также формируют гидрофобные оболочки клеточных органоидов и мембран. За счет гидрофобных (в основном дисперсионные, а также, отчасти, индукционные) связей неполярные участки радикалов аминокислот в структуре белка и радикалов нуклиотидов в структуре нуклеиновых кислот, радикалы липидов в липидных оболочках и т.п. располагаются упорядоченно; а не создают неопределенность положения в молекуле и органоиде вцелом, свободно изгибаясь и мешая работе организма.
Другой важной особенностью дисперсионного взаимодействия является его аддитивность. Например, если имеются три частицы, то общая энергия взаимодействия U>123> слагается из энергий попарного их взаимодействия U>12>, U>23> и U>31>: U>123 >= U>12>+U>13>+U>31>. Наглядно аддитивность дисперсионных взаимодействий можно объяснить как результат согласованного в такт движения электронных осцилляторов, понижающего общую энергию нулевых колебаний системы. Аддитивность дисперсионных сил проявляется в адсорбции и других процессах связанных с конденсацией газа.
Дисперсионные силы играют большую роль при взаимодействии не только отдельных молекул, но и коллоидных частиц. Благодаря аддитивности дисперсионных сил энергия взаимодействия одной молекулы коллоидной частицы со всеми молекулами другой коллоидной частицы убывает уже не пропорционально шестой, а только третьей степени расстояния.
Если же учесть взаимодействие всех
молекул одной коллоидной частицы со
всеми молекулами другой коллоидной
частицы, то энергия дисперсионного
взаимодействия, отнесенная к единице
поверхности, спадает пропорционально
квадрату расстояния между поверхностями
макрочастиц:
 ,
где s - расстояние
между поверхностями частиц; n
- число молекул в единице объема
вещества; I>1>,
I>2> - потенциалы
ионизации частиц; >1>,
>2> - величины
поляризуемости частиц. Таким образом,
суммарное взаимодействие оказывается
значительным и на больших расстояниях.
Исследование этого эффекта советскими
физиками Ландау и Лифшицем и изучение
Дерягиным сил отталкивания между
коллоидными частицами позволило
Дерягину разработать современную
теорию устойчивости и коагуляции
коллоидных систем.
,
где s - расстояние
между поверхностями частиц; n
- число молекул в единице объема
вещества; I>1>,
I>2> - потенциалы
ионизации частиц; >1>,
>2> - величины
поляризуемости частиц. Таким образом,
суммарное взаимодействие оказывается
значительным и на больших расстояниях.
Исследование этого эффекта советскими
физиками Ландау и Лифшицем и изучение
Дерягиным сил отталкивания между
коллоидными частицами позволило
Дерягину разработать современную
теорию устойчивости и коагуляции
коллоидных систем.
Особый вид дисперсионных взаимодействий наблюдается в, так называемых, лиофильных коллоидах (в истинных растворах высокомолекулярных соединений). У молекул, составляющих лиофильные коллоидные системы, длина осциллирующего диполя так велика, что взаимодействуют, по сути, не мгновенные диполи, а меняющие положение отдельные заряды; причем у каждой молекулы высокомолекулярного соединения, составляющего лиофильную коллоидную систему, одновременно существует множество положительных и множество отрицательных полюсов. Это, названное Лондоном униполярным, взаимодействие существенно в молекулах с сопряженными связями, в высокополимерных и т.п. соединениях, где электрон может перемещаться вдоль цепочки сопряженных связей. Энергия униполярного взаимодействия обратно пропорциональна второй степени расстояния.
Ван-дер-ваальсово отталкивание
(эффект Паули)
Выше были описаны три основных типа дальнодействующих сил ответственных за ван-дер-ваальсовое притяжение между молекулами: эффекты Лондона, Кезома и Дебая. При сближении молекул (или их частей), наряду с вышеизложенными дальнодействующими силами, заметными становятся также короткодействующие силы, возникающие при перекрывании электронных облаков молекул (или частей молекул). На больших расстояниях эти силы несущественны, так как электронная плотность спадает практически до нуля уже на отдалении порядка 3Å от ядра атома.
Перекрывание электронных облаков может привести ко двоякого рода результатам: если у частиц имеются незаполненные целиком или низко лежащие свободные молекулярные орбитали, могут образоваться донорно-акцепторные, координационные, межмолекулярные и другие химические соединения; короткодействующие силы другого вида — силы ван-дер-ваальсового отталкивания, возникающие при перекрывании полностью заполненных атомных или молекулярных электронных оболочек, связанных с проявлением принципа Паули.
Принцип Паули (принцип исключения Паули, запрет Паули) играет фундаментальную роль в поведении многоэлектронных систем. Согласно принципу Паули на одной спин-орбитали не может находиться двух электронов с одинаковым набором четырех квантовых чисел. Принцип исключения Паули отностся к основным законам природы и выражает одно из важнейших свойств не только электронов, но и всех других обладающих полуцелыми значениями спинового квантового числа микрочастиц (в том числе: протонов, нейтронов, многих других элементарных частиц, а также многих атомных ядер).
Например: если две молекулы H>2> в основном состоянии оказываются очень близко друг к другу, между ними возникают силы отталкивания: два электрона первой молекулы на орбитали σ1s и два электрона второй молекулы на такой же σ1s-орбитали оказываются в одной области пространства; но так как в граничной области σ1s-орбитали может находиться не более чем два электрона с антипараллельными спинами (запрет Паули), то обе пары электронов двух столкнувшихся молекул будут стремиться отдалиться друг от друга. Электронная плотность в области соприкосновения понизится, и кулоновское отталкивание отделит одну пару ядер от другой; слияние системы в молекулу H>4> не произойдет. В ван-дер-ваальсовом отталкивании проявляется насыщаемость химических связей.
Аналогичное состояние возникает при сближении и других молекул, орбитали которых заполнены парами электронов с антипараллельными спинами: при перекрывании молекулярных орбиталей возникает эффект ван-дер-ваальсового отталкивания (иногда силы отталкивания, возникающие при перекрывании полностью заполненных электронных оболочек, назавают "силами Паули"). Природа их, также как и других сил Ван-дер-Ваальса – электрическая.
Силы ван-дер-ваальсового отталкивания — важнейшая компонента межмолекулярного взаимодействия. На коротких расстояниях они значительны и возрастают при сближении очень быстро. Энергию межмолекулярного отталкивания аппроксимируют обычно одним из следующих выражений:
 или
или  .
.
A и ρ (или B и n) – константы, определяемые при столкновении атомов инертных газов и простейших молекул. Обычно n=12.
§2 Водородные связи

В тех случаях, когда водород соединен с сильно электроотрицательным элементом, он может образовать еще одну дополнительную связь (редко две; больше, - почти никогда), правда, как правило значительно менее прочную, чем обычная валентная связь (единственным исключением является фтороводород, в его структуре водородная связь неотличима от химической и частично сохраняется даже в газообразном состоянии). Способность атома водорода связывать в ряде случаев два атома впервые была установлена в восьмидесятых годах прошлого столетия М.А. Ильинским и Н.Н. Бекетовым.
Водородные связи по силе взаимодействия занимают промежуточное положение между химическим и ван-дер-ваальсовым взаимодействиями. Условно водородные связи относят к межмолекулярным, однако во многих высокомолекулярных типичные неметаллы содержащих органических соединениях (например в белках, в нуклеиновых кислотах) водородные связи являются внутримолекулярными и формируют вторичную структуру этих молекул. Химические соединения, содержащие водород непосредственно связанный с типичными неметаллами (F, O, N, Cl, S), имеют по сравнению с соединениями похожего строения, но в которых водород связан с другими элементами, либо с типичными неметаллами связан не водород, существенно более высокие температуры кипения и сублимации, а энергия на их сублимацию или испарение уходит значительно большая. Считается, что причиной повышенных температур и удельных теплоемкостей сублимации и кипения водородсодержащих соединений типичных неметаллов является то, что они находятся в конденсированном состоянии не в виде мономеров, а в виде олигомеров (ассоциатов), причем полимеризация мономеров происходит посредством водородных, а не химических, связей.
Водородная связь занимает промежуточное положение между химической и физической связями также и по своей природе. Эта связь обусловлена тем, что смещение электрона от атома водорода превращает его в частицу с уникальными свойствами. Если рассматривать эту частицу как катион, то она: во-первых не имеет электронов и поэтому в отличие от других катионов не отталкивается электронными оболочками от других частиц, а испытывает только притяжение, и во-вторых обладает ничтожно малым размером (протон и дейтрон в тысячи раз меньше остальных ионов).
Водородная связь проявляется тем сильнее, чем больше электроотрицательность атома-партнера, и чем меньше его размеры; поэтому она характерна прежде всего для соединений фтора, а также кислорода, в меньшей степени азота, в еще меньшей степени для хлора и серы соответственно. Соответственно меняется и энергия водородной связи. В общем случае она зависит как от вида и состояния атома-партнера, так и от того, с какими атомами последний соседствует. Так, энергия водородной связи H……F (Здесь и вдальнейшем эта связь обозначена точками) равна ≈10 ккал, связи H……O ≈5 ккал, связи H……N ≈2 ккал. Соседство сильно электроотрицательных атомов (F, O, N, Cl) может активизировать к образованию водородной связи и атомы CH групп (хотя электроотрицательности углерода и водорода почти одинаковы). Этим объясняется объясняется возникновение водородных связей в таких, например, соединениях, как альдегиды, хлорсодержащие и фторсодержащие углеводороды, содержащие нитрогруппу органические соединения и др.
С усилением водородной связи уменьшаются
и соответствующие расстояния. Так если
в структуре конденсированного
аммиака расстояние H……N
превышает расстояние N—H
значительно более чем вдвое то в
структуре воды (и льда) расстояние H……O
больше расстояния O—H
лишь на ≈70%, а для
фтороводорода оба расстояния (F—H
и H……F)
одинаковы, т.е. протон располагается
строго по середине между ионами фтора.
усилением водородной связи уменьшаются
и соответствующие расстояния. Так если
в структуре конденсированного
аммиака расстояние H……N
превышает расстояние N—H
значительно более чем вдвое то в
структуре воды (и льда) расстояние H……O
больше расстояния O—H
лишь на ≈70%, а для
фтороводорода оба расстояния (F—H
и H……F)
одинаковы, т.е. протон располагается
строго по середине между ионами фтора.
Б лагодаря
водородным связям молекулы объединяются
в ассоциаты (димеры или полимеры).
Последние могут иметь линейное,
разветвленное или кольчатое строение.
Так, муравьиная кислота как в жидкой,
так и в газообразной фазе существует
главным образом в виде димера;
его структура установлена методом
электронографии (см. рис.3). Способность
к ассоциации отличает воду, спирты, и
многие другие жидкости от неассоциированных
жидкостей (например от углеводородов).
Ассоциация приводит к повышению
температуры плавления, температур
сублимации и кипения, теплоты
парообразования, а также к изменению
взаимной растворяющей способности
веществ.
лагодаря
водородным связям молекулы объединяются
в ассоциаты (димеры или полимеры).
Последние могут иметь линейное,
разветвленное или кольчатое строение.
Так, муравьиная кислота как в жидкой,
так и в газообразной фазе существует
главным образом в виде димера;
его структура установлена методом
электронографии (см. рис.3). Способность
к ассоциации отличает воду, спирты, и
многие другие жидкости от неассоциированных
жидкостей (например от углеводородов).
Ассоциация приводит к повышению
температуры плавления, температур
сублимации и кипения, теплоты
парообразования, а также к изменению
взаимной растворяющей способности
веществ.
Как уже указывалось, энергия водородной связи обычно значительно меньше энергии ковалентной связи. Поэтому повышение температуры приводит к разрыву водородных связей; однако этот процесс как правило, растянут на сравнительно широкий интервал температур; в карбоновых кислотах, например, ассоциация сохраняется даже при их парообразовании. Следует однако заметить, что при более существенном повышении температуры разрушаться начинают и донорно-акцепторные, а также ковалентные связи, разрушение химических связей является основной возможной причиной образования, так называемой, горячей плазмы.
Если взять ряд сходных веществ, то с ростом молекулярного веса следует ожидать увеличения их температур плавления и кипения, а также теплот парообразования; однако как видно из табл. 2, при переходе от HF к HCl и от H>2>O к H>2>S происходит резкое падение численного значения указаных свойств.
Таблица 2
Температура плавления, температура кипения и теплота парообразования (в точке кипения)
для гомологов воды и фтороводорода.
|
Вещество |
Температура плавления, oC |
Температура кипения, oC |
Теплота парообразования, ккал/моль |
|
H>2>O |
0,0 |
100,0 |
9,75 |
|
H>2>S |
-85,5 |
-60,7 |
4,5 |
|
H>2>Se |
-64,8 |
-41,5 |
5,1 |
|
H>2>Te |
-49,0 |
-2,0 |
5,8 |
|
HF |
-83,1 |
-19,5 |
7,2 |
|
HCl |
-112,0 |
-84,9 |
3,6 |
|
HBr |
-87,0 |
-66,8 |
3,9 |
|
HI |
-50,9 |
-39,4 |
4,2 |
Это объясняется тем, что между молекулами HF и между молекулами H>2>O существуют сильные водородные связи. Масштаб этого эффекта виден из рис. 4.
Наиболее удобным индикатором водородной связи является температура кипения, т.к. ее легко измерить. Определив температуру какого-либо спирта ROH и соответствующего ему меркаптана RSH, можно убедиться, что для ROH она больше, чем для RSH. Простые эфиры даже с большим молекулярным весом, чем спирты, более летучи. Если бы вода не была ассоциированной жидкостью, то она имела бы температуру плавления около -100oC и температуру кипения около -80oC (что легко установить с помощью чертежа подобного рис.4).
Е сли
воспользоваться методом сравнительного
расчета и сопоставить температуру
кипения в рядах HЭ (Э = F,
Cl, Br, I) и H>2>Э
(Э = O, S, Se, Te) (рис.
5), то из характера отклонения точки
(HF; H>2>O)
и из того факта, что молекулы водяного
пара почти не ассоциированы, можно
заключить, что в отличие от воды
ассоциация фтороводорода сохраняется
и в паровой фазе (в противном случае
следовало бы ожидать расположения на
прямой всех четырех точек); это
свидетельствует о большей прочности
связи H……F
по сравнению со связью H……O.
Cделанный вывод можно подтвердить
и заметно меньшей разницей в теплотах
парообразования HF и
HCl по сравнению с разницей в теплотах
парообразования H>2>O
и H>2>S
(см. табл. 2). Действительно, в парах
фтороводорода существуют молекулы
(HF)>n>
, имеющие следующую структуру:
сли
воспользоваться методом сравнительного
расчета и сопоставить температуру
кипения в рядах HЭ (Э = F,
Cl, Br, I) и H>2>Э
(Э = O, S, Se, Te) (рис.
5), то из характера отклонения точки
(HF; H>2>O)
и из того факта, что молекулы водяного
пара почти не ассоциированы, можно
заключить, что в отличие от воды
ассоциация фтороводорода сохраняется
и в паровой фазе (в противном случае
следовало бы ожидать расположения на
прямой всех четырех точек); это
свидетельствует о большей прочности
связи H……F
по сравнению со связью H……O.
Cделанный вывод можно подтвердить
и заметно меньшей разницей в теплотах
парообразования HF и
HCl по сравнению с разницей в теплотах
парообразования H>2>O
и H>2>S
(см. табл. 2). Действительно, в парах
фтороводорода существуют молекулы
(HF)>n>
, имеющие следующую структуру:
И хотя для большинства частиц n=4, но есть и частицы, для которых n=5 и 6.
Рассмотренные выше примеры относились случаю межмолекулярной водородной связи, но нередко водородная связь объединяет части одной и той же молекулы, т.е. является внутримолекулярной. Это характерно для многих органических веществ; именно посредством водородных, а не химических, ионных или ван-дер-ваальсовых связей формируются вторичная структура белков, а также структура гидратных оболочек у минеральных солей и органических веществ. Водородные связи играют основную роль в формировании также третичной и четвертичной структуры глобулярных белков. Предполагают, что действие памяти связано с хранением информации в конфигурациях с водородными связями.
В большинстве случаев образования внутримолекулярной водородной связи водород входит в плоское шестичленное кольцо; если возникновение такого цикла затруднено, то внутримолекулярная водородная связь обычно не образуется. Вот несколько примеров образования внутримолекулярной водородной связи:
Е
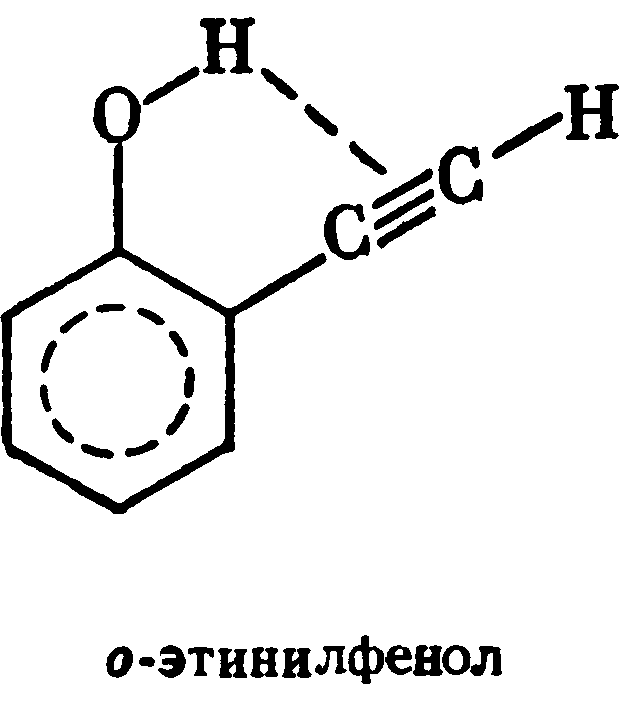

сли
у о нитрофенола водородная связь –
внутримолекулярная, то у n нитрофенола
она межмолекулярная, так как в последнем
веществе водород удален от кислорода
нитрогруппы. Константа
диссоциации 2, 6 диоксибензойной
кислоты при 25oC
равна 5·10-2, для 3, 5 диоксибензойной
кислоты при 25oC
она в 550 раз меньше. Это объясняется
тем, что в 3, 5 диоксибензойной
кислоте внутримолекулярная водородная
связь почти не проявляется, что усиливает
связь O—H в карбоксильной
группе. Молекула о этинилфенола
особенно интересна тем, что в ней
водородная связь образуется за счет
π электронов тройной связи.
У чет
влияния водородной связи позволил
осмыслить многие факты. Так, образование
солей типа KHF>2 >и NaHF>2>
объясняется существованием прочного
иона HF>2>‾‾,
образующегося в результате процесса
чет
влияния водородной связи позволил
осмыслить многие факты. Так, образование
солей типа KHF>2 >и NaHF>2>
объясняется существованием прочного
иона HF>2>‾‾,
образующегося в результате процесса
 ;
действительно, при н.у. равновесие
;
действительно, при н.у. равновесие
 смещено вправо (K>298>=5,1);
энергия водородной связи в F—H……F–
составляет 27 ккал/моль. Влияние
водородной связи делает понятным и то
обстоятельство, что фтороводородная
кислота в отличие от ее аналогов (HCl,
HBr, HI) не является сильной кислотой;
ее константа диссоциации равна 7,2·10-4.
смещено вправо (K>298>=5,1);
энергия водородной связи в F—H……F–
составляет 27 ккал/моль. Влияние
водородной связи делает понятным и то
обстоятельство, что фтороводородная
кислота в отличие от ее аналогов (HCl,
HBr, HI) не является сильной кислотой;
ее константа диссоциации равна 7,2·10-4.
Важную роль водородные связи играют в структуре воды и льда. На рис. 6 показан фрагмент структуры льда Ι. Каждый атом кислорода в этой структуре тетраэдрически связан с четырьмя другими атомами кислорода, между ними располагаются атомы водорода; два атома водорода соединены с данным атомом кислорода полярной ковалентной связью (d=1,011Ǻ), два других — водородной связью (d=1,752 Ǻ; E>O>…>H> ≈ 5ккал/моль), т.е. входят в состав двух других молекул H>2>O. Создается ажурная структура, далекая от плотной упаковки. Отсюда небольшая плотность и значительная рыхлость льда. При плавлении льда водородные связи частично разрушаются (примерно на 10%); это несколько сближает молекулы, поэтому вода немного плотнее льда. Нагревание воды, с одной стороны, приводит к ее расширению, т.е. к увеличению объема, с другой стороны, оно вызывает дальнейшее разрушение водородных связей и тем самым уменьшение объема. В результате плотность воды проходит через максимум (при 4oC).
Водородная связь играет большую роль в процессах растворения, так как растворимость зависит от способности вещества создавать водородные связи с растворителем; при этом часто образуются продукты их взаимодействия — сольваты. В качестве примера можно оказать на растворение спиртов в воде. Этот процесс сопровождается выделением теплоты и уменьшением объема, т.е. признаками соответствующими образованию соединений. Отсутствием влияния водородной связи можно объяснить и те случаи, когда полярные соединения нерастворимы в воде. Так, полярный йодистый этил хорошо растворяет неполярный нафталин, а сам не растворяется в таком полярном растворителе, как вода.
Вопрос о природе водородной связи окончательно не решен. Ясно, что здесь играют роль и междипольное взаимодействие, и эффект поляризации, и донорно-акцепторный механизм. Трудность квантово-механического расчета водородной связи обусловлена тем, что погрешность вычисления значительно больше величины энергии водородной связи. По-видимому наиболее надежные результаты можно ожидать от метода молекулярных орбиталей.
Водородная связь встречается почти повсеместно — и в органических кристаллах (содержащих атомы C, H и O), и в белках (в них есть атомы C, H и N), и в полимерах, а следовательно живые организмы всегда изобилуют водородными связями. Предполагают, что и действие памяти связано с хранением информации в конфигурациях с водородными связями. "Всеобщность" водородной связи обусловлена также тем, что молекулы H2O встречаются повсеместно, а каждая из них, имея в своем составе два атома водорода и две необобщенные электронные пары, может образовать четыре водородные связи.
Выводы
Из всего рассмотренного материала можно заключить, что между физическими связями и химическими связями нельзя провести четкую границу. Физические и химические связи имеют общую физическую природу (электрическую). Кроме того, один и тот же эффект (эффект исключения Паули) считается поддерживающим устойчивость химической связи в пределах молекулы (или ее участка) либо устойчивость физической связи в пределах фазы только в зависимости от того считаются ли взаимодействующие атомы связанными химически или несвязанными химически. По длине и энергии связи физические и химические связи тоже граничат вплотную.
Единственным фактором который позволяет отделить физические связи химических может быть лишь насыщаемость/ненасыщаемость связи. Однако, и в этом случае разделение связей на химические и физические будет нестрогим вследствие существования водородной (иногда ее также называют протонной) связи, насыщаемость которой нечетко выражена (вследствие того, что водородная связь является переходной между различными механизмами химического и межмолекулярного взаимодействия практически по всем параметрам ее уже сейчас часто относят как к первым так ико вторым). Кроме того, такое разделение связей на физические и химические требует отнесения также ионной связи к физическим, а не к химическим связям, вследствие ее явной ненасыщаемости.
Литература
Краснов К.С., Молекулы и химическая связь, "Высшая школа", Москва 1984.
Карапетьянц М.Х., Дракин С.И., Строение вещества, "Высшая школа", Москва 1978.
Зацепина Г.Н., Физические свойства и структура воды, МГУ, Москва 1998.
Кнорре Д.Г., Крылова Л.Ф., Музыкантов В.С., Физическая химия, "Высшая школа", Москва 1990.
Мушкамбаров Н.Н., Аналитическая биохимия, "Экспедитор", Москва 1996.
Николаев А.Я., Биологическая химия, ООО "Медицинское информационное агентство", Москва 1998.