Методы химического анализа
Введение в учебную дисциплину
Рыночная экономика не может обходиться без широкого использования современных методик по оценке производственной и рыночной новизны, технического уровня и конкурентоспособности производимой и продаваемой продукции. Только в этом случае возможно успешное регулирование уровня качества выпускаемой продукции на предприятиях в рамках систем качества.
Особые требования предъявляются к методическим основам оценки качества продукции при подготовке и проведении обязательной и добровольной сертификации, которые должны наиболее полно обеспечивать:
возможность выявления, комплексного анализа и достоверной оценки всей совокупности потребительских свойств, безопасности и экологичности продукции;
создание на основе проведенной оценки предпосылок для социальной защиты потребителей от функционально и экологически опасной продукции, а также от несоответствия реальной цены на продукцию её качеству.
Выполнение требований может быть достигнуто комплексным решением технических и управленческих задач, к которым относятся:
1. использования современных методик по оценке производственной и рыночной новизны, технического уровня и конкурентоспособности производимой и продаваемой продукции а также объективное отражение этих свойств и показателей в нормативно-технической документации на продукцию и в результатах оценки её качества.
2. Оценка качества продукции (в любых формах представления её результатов) на различных этапах взаимодействия разработчиков, изготовителей и потребителей с учётом взаимосвязи качества, количества и цены потребления.
3. Оперативное получение необходимых данных об уровне качества продукции и её конкурентоспособности на любом этапе «петли качества».
4. Своевременное применение руководством предупреждающих и корректирующих действий для совершенствования систем и процессов разработки, изготовления и реализации продукции.
Решение перечисленных задач возможно только при наличии достаточного количества зарегистрированных и доступных для измерения показателей, отражающих уровень качества продукции. Без них невозможна выработка необходимых управляющих воздействий в системе управления качеством продукции.
Теории измерения показателей, создаваемых или используемых человеком продукции и процессов, разрабатываются различными науками. Исследование как количественных, так и неколичественных методов и оценка уровня качества продукции осуществляется с использованием методов межотраслевой (междисциплинарной) науки квалиметрии, её задачи:
определение номенклатуры и численных значений показателей качества для включения их в техническое задание на разработку и нормативную документацию с целью последующего контроля и сопоставительной оценки с продукцией аналогичного назначения;
формирование требований к показателям качества для включения их в нормативную документацию (стандарты, технические регламенты и условия);
оценка качества продукции на основе полученных показателей в ходе её испытаний и контроля;
разработка стратегий обслуживания технических устройств на основе данных о показателях надежности.
Особое место в решении задач квалиметрии по формированию системы и регистрации показателей качества отводится физическим и физико – химическим методам аналитического контроля. Применение их обеспечивает проведение качественного и количественного анализа веществ, входящих в состав выпускаемой продукции. Результатами анализа являются полученные параметры веществ,
отражающие их состав и свойства, без которых выводы и заключения о качестве продукции могут быть весьма спорными. Роль и значение параметров состава и свойств веществ для различных видов продукции раскрываются в последующих вопросах настоящей темы.
Формирование и регистрация параметров веществ, входящих в состав продукции, осуществляется на всех стадиях её жизненного цикла ( рис.1):
1. Исследования и обоснования разработки продукции. В ходе проведения научно – исследовательских работ обосновываются ожидаемые и достижимые показатели качества.
2. Разработки изделия или технологии производства материала. При проведении опытно – конструкторских и лабораторно – исследовательских работ уточняются показатели качества и включаются в технологическую документацию на производство продукции.
3. Производства продукции. На предприятиях организуется проверка выпускаемой продукции, в ходе которой контролируются, уточняются и совершенствуются показатели качества, а при необходимости вносятся корректирующие изменения в технологическую документацию на её производство.
4. Эксплуатации продукции. Эта стадия отличается от остальных реализацией показателей качества при вводе изделий в эксплуатацию, хранении и транспортировании, использовании их по назначению и поддержания в исправном состоянии – обслуживании. В ходе каждого из перечисленных этапов используются определенные показатели, отражающие начальное состояние изделия, его характеристики и возможные изменения при нарушении условий эксплуатации. Показатели качества, претерпевшие изменения при эксплуатации продукции, как правило, обрабатываются, анализируются и применяются в качестве исходных данных при совершенствовании или создании новой продукции.









Хранение
Транспортирование


Использование
по
назначению
Поддержание в исправном состоянии

Обслуживание
Ремонт

Утилизация
Рис.1. Стадии жизненного цикла изделий
5. Восстановления работоспособности и исправности изделий. Эта стадия характеризует неработоспособное состояние изделий, требующих ремонта и восстановления ресурса. До перехода изделия в эту стадию его показатели качества, как правило, изменяются в сторону уменьшения первоначальных значений, по причинам естественного старения и износа материалов. Для определения технического состояния изделий применяются диагностические показатели, которые близки по содержанию показателям качества.
На любой из стадий жизненного цикла продукции возникает необходимость контроля качества материалов, так как от их состояния существенно зависят потребительские и эксплуатационные свойства изделий, а также безопасность их использования по назначению. Поэтому специалисты по качеству должны в совершенстве владеть методами анализа состава продукции и измерения её параметров. Для этого необходимо изучить соответствующие методики и освоить приёмы использования современных приборов, применяемых в лабораторных условиях и в производственной практике.
Утилизации подлежит продукция, потерявшая свои свойства в процессе эксплуатации или дальнейшее использование которой нецелесообразно. Применяются различные способы утилизации (переработка, сжигание, захоронение и др.), в результате образуются вторичные продукты, которые не всегда безвредны и нередко наносят существенный вред окружающей среде. Параметры последствий воздействия продукции на окружающую среду после её утилизации и методы их контроля должны разрабатываться в ходе исследовательских и опытно-конструкторских работ.
Учебная дисциплина «Физико – химические методы аналитического контроля» выполняет функцию специального раздела в ряду дисциплин (метрология, стандартизация, сертификация и управление качеством), изучающих правила управления качеством и подтверждения качества продукции. Наиважнейшей задачей учебной дисциплины является ознакомить студентов с современными методами анализа продукции, которые наиболее широко используются для решения текущих химико – технологических задач производства и проверки качества продукции.
На изучение дисциплины отводится 66 часов учебного времени, из них для лекционных занятий – 32 часа и выполнение лабораторных работ – 34 часа. Лабораторные работы будут проведены в соответствии с разработанным на кафедре лабораторным практикумом, должны быть выполнены каждым студентом и обязательно защищены. Завершается изучение дисциплины сдачей экзамена.
Литература, рекомендованная для изучения
1.М.В.Кулаков.Технологические измерения и приборы для химических производств.Изд. Москва «Машиностроение» 1983. . Библиотека ТГТУ инв. .№ Л 11 / К 90
2. В. А. Прохоров. Основы автоматизации аналитического контроля химических производств. Изд. Москва «Химия»1984. Библиотека ТГТУ инв. .№ Л 11 / П 844.
3.Г. Юинг. Инструментальные методы химического анализа. Изд. Москва «Мир» 1989. Библиотека ТГТУ инв. .№ Г 4 / Ю 22.
4.Аналитическая химия. Физические и физико – химические методы анализа. Под ред. проф. Петрухина. Изд. Москва «Химия» 2001. Библиотека ТГТУ инв. .№ Г 4 / А 64. Библиотека ТГТУ инв. .№ Л 11 / П 844.
5. В.Ф.Барковский, Физико-химические методы анализа. Изд. Москва «Высшая школа» 1983 г. Библиотека ТГТУ инв. .№ Г 4 / 252.
6. В.П.Васильев. Аналитическая химия. Физико – химические методы анализа, ч. 2. Изд. Москва «Высшая школа» 1989.
7. Б. И. Герасимов. Микро процессорные аналитические приборы. Изд. Москва «Машиностроение» 1989 г. Библиотека ТГТУ инв. .№К 9 / Г 371.
1. Теоретические основы аналитического контроля качества продукции
Обеспечение качества продукции представляет собой многоплановый процесс, включающий проведение комплекса мероприятий по формированию, контролю и поддержанию характеристик, отражающих свойства продукции Наиважнейшим мероприятием является аналитический контроль, который проводится на всех стадиях жизненного цикла продукции. В зависимости от решаемых задач при проведении аналитического контроля применяются различные методы для определения состава и измерения параметров продукции.
1.1 Общие сведения о методах анализа состава и измерения параметров продукции
В основе определения состава и свойств продукции лежит химический анализ. Он связан с проведением качественного и количественного анализа состава продукции и сравнением полученных результатов с установленными нормативно-технической документацией требованиями.
Химический анализ в широком смысле этого понятия, включающий физические и физико-химические методы, является составной частью метрологии. Его особенностью является предварительное проведение качественного анализа, т. е. идентификации химических частиц различного рода (атомов, молекул, ионов, радикалов) с последующим определением их количества (качественный анализ) в анализируемом продукте.
Цели, с которыми проводится качественный или количественный химический анализ состава продукции разнообразны. В зависимости от решаемых задач и глубины проверки продукции результаты могут быть получены проведением следующих анализов: атомного, молекулярного, функционального и валового. Атомный (элементный) и молекулярный анализы заключаются в контроле состава веществ на уровне атомов или молекул. Функциональный анализ заключается в определении состава функциональных групп в химических соединениях. Валовой анализ применяется в случае проверки сложных смесей веществ (горные породы, цемент), когда состав пробы выражается в виде условно выбранных соединений, например оксидов.
Состав продукции проверяется измерением количества или физических свойств, входящих в неё веществ. Измерения производятся непосредственно или же после соответствующей подготовки продукции (разделение, концентрирование, перевод в удобную для измерения форму и др.). Процесс завершается измерением величины аналитического сигнала. Для получения аналитического сигнала, как правило, используются три группы методов: химические, физические и физико-химические.
Химические методы основаны на химических реакциях определяемого компонента с реагентом. Эффектом реакции может быть образование малорастворимого осадка, малодиссациированного соединения или прочного комплексного соединения.
В физических методах измеряется свойство (интенсивность излучения света, радиоактивного излучения и др.), непосредственно зависящее от природы атомов и их концентрации в веществе. При этом химические реакции или совсем не играют роли, или имеют второстепенное значение.
В физико-химических методах анализа определяются изменения физических свойств системы (коэффициента преломления света, электрической проводимости, поглощения света и др.), происходящие в результате химических или электрохимических реакций. Интенсивность физического сигнала зависит от концентрации определяемого компонента.
Между химическими и физико-химическими, физическими и физико-химическими методами анализа не всегда можно провести чёткую границу. Например, измерение электрической проводимости растворов (кондуктометрия) не требует проведения химических реакций и относится к физическим методам, тогда как определение изменения электрической проводимости при титровании кислоты щёлочью (кондуктометрическое титрование) является физико-химическим методом. Иногда физические и физико-химические методы объединяются под общим названием инструментальные методы, так как для измерения сигналов используется прецизионная аппаратура.
1.2. Физико- химические методы анализа и их место в системе контроля качества продукции
Свойства веществ и материалов, производимой и реализуемой продукции, изучаются с использованием методов современной аналитической химии, которые направлены на решение задач управления качеством продукции.
Основными рабочими средствами аналитической химии являются физические и физико – химические методы анализа. Всё большее число используемых в них принципов контроля реализуются в инструментальных методах. Появляются узкоспециализированные приборы для автоматического контроля химико – технологических процессов. Увеличивается число приборов, сочетающих несколько аналитических методов (газовые и жидкостные хроматографы, хромато-масс-спектрометры и др.).
Физические и физико-химические методы анализа являются естественным продолжением курса химических методов анализа, и основывается на регистрации аналитических сигналов, появление которых зависит от физико-химических свойств вещества, его природы и содержания в анализируемом продукте.
Классические методы анализа применяются в специализированных аналитических лабораториях. Их проведение связано с периодическим отбором проб анализируемых продуктов, что не всегда удобно, эффективно и не обеспечивает высокую скорость получения результата. Вместе с тем, они не в состоянии удовлетворить многообразные запросы науки, техники, промышленности и социальной жизни людей. Этих недостатков лишены физические и физико-химические методы, а доступность аппаратуры делает их востребованными в практике всех сфер деятельности людей.
Современные отрасли производства и социальной жизни людей ставят свои специфические задачи перед физическими и физико – химическими методами анализа по контролю качества продукции.
Выплавляя чугун или сталь, металлург должен знать качественный и количественный состав плавок. Вместе с содержанием основного металла в сплаве ему необходимы данные о составе используемых исходных веществах и их свойствах. Контроль этих параметров позволяет непосредственно судить о режиме плавки, так как они характеризуют качество получаемых сплавов, а также при необходимости производить соответствующие корректировки технологических процессов. Например, жаропрочные сплавы металлов теряют свои свойства, если количество “запрещенных” примесей в них превысит 10-5%. Вместе с тем, определение малых концентраций примесей практически невозможно химическими методами. Поэтому для решения задач такого рода применяются физические и физико-химические методы анализа, обладающие самым низким пределом обнаружения примесей.
В ходе протекания химико-технологических процессов производства продукции изменяются химический состав перерабатываемых веществ и их свойства. Контроль этих параметров позволяет непосредственно судить о режиме процесса, составе получаемых продуктов, а скорость получения данных своевременно вносить соответствующие корректировки. Поэтому на химических предприятиях применяются методы автоматизированного контроля, которые реализуются с применением приборов называемых анализаторами.
Наряду с черной и цветной металлургией, химической промышленностью и другими традиционными отраслями большое значение стали иметь отрасли по освоению атомной энергии в мирных целях, связанные с ракетостроением, освоением космоса, развитием полупроводниковой промышленности, электроники, компьютеров, чистых и сверхчистых веществ.
Развитие перечисленных отраслей поставило перед специалистами задачу снизить предел обнаружения примесей в производимых веществах до 10-5 – 10-10%.Это стало возможным только при условии применения физических и физико-химических методов анализа.
Впечатляют примеры, показывающие связь свойств с загрязнением примесями полупроводниковых материалов, из которых изготавливаются радиоэлектронные элементы с загрязнением исходных материалов, используемых для их изготовления «вредными» примесями. Германий, применяемый в электронной промышленности, утрачивает свои полупроводниковые свойства, если загрязнен фосфором или мышьяком в пределах 10-10%. Цирконий, являющийся конструкционным материалом для ядерной промышленности, при наличии в нем примеси гафния в пределах 10-5%, недопустим к применению.
Подобные примеры можно приводить и с лекарственными препаратами, продукцией парфюмерной, пищевой и текстильной промышленности. Наличие вредных примесей в них может негативно повлиять на состояние здоровья людей. Поэтому без применения физических и физико-химических методов анализа сложно контролировать выпуск продукции, проверить качество поступившей в продажу продукции, а значит и разрешать возникающие спорные вопросы между покупателем и продавцом.
Особенное значение приобрели физико-химические методы анализа для решения задач экологической направленности, а также в медицинской и судебно-экспертной практике, так как только с их помощью можно быстро получить достоверные результаты.
Нельзя обойти стороной применение физических и физико-химических методов анализа в военном деле и гражданской обороне. Методы, реализованные в средствах радиационной, химической и биологической разведки позволяют оперативно проводить проверку зараженности атмосферы, техники, имущества, продуктов питания и идентифицировать токсичные вещества. Войсковые газоанализаторы позволяют определять в атмосфере токсичные вещества в концентрациях до 10-5%. Индикаторы для определения сильнодействующих ядовитых веществ (СДЯВ, табл. 1) и токсичных примесей в испарениях ракетного топлива реагируют на концентрации10-5–10-7%, что многократно превышает предельно-допустимые нормы.
Таблица 1
Предельно допустимые нормы концентраций
сильнодействующих ядовитых веществ в атмосфере
|
№ п/п |
Наименование СДЯВ |
Величина пороговой токсодозы, г/см3 |
|
1 |
Аммиак |
454 |
|
2 |
Гидразин |
14 |
|
3 |
Окись углерода |
1620 |
|
4 |
Окись этилена |
3600 |
|
5 |
Двуокись серы |
194 |
|
6 |
Сероводород |
2592 |
|
7 |
Фосген |
13 |
|
8 |
Цианистый водород |
36 |
|
9 |
Хлор |
36 |
Примечание. В таблице приведены значение пороговых токсодоз для взрослых людей, для детей – в 4-10 раз меньше.
Важной задачей физических и физико-химических методов анализа является также разработка экспресс методов обнаружения и количественного определения отдельных элементов в составе выпускаемой продукции. Всё перечисленное активизировало развитие аналитического приборостроения, инициировало разработку методов автоматизации контроля химико - технологических процессов, связанных с производством продукции и обеспечения безопасности жизнедеятельности людей. Современное лабораторное аналитическое оборудование позволяет быстро выявить изменения в продукции предназначенной для длительного хранения или, хранящейся с нарушением установленных требований, а также разрешить возникающие спорные вопросы между производителем и потребителем.
1.3 Классификация физико-химических методов анализа
К наиболее востребованным в научной, производственной и социальной практике физическим и физико-химическим методам относятся спектральные, электрохимические и хроматографические методы анализа, рис.2. Они отличаются большим разнообразием, как по принципу действия, так и по технике исполнения в пределах каждого метода и для их изучения потребуется значительно больше времени, чем выделено для учебной дисциплины. Поэтому на занятиях будут рассмотрены приемы лишь тех методов, которые нашли наиболее широкое применение на практике, а также изучены устройства и приборы, используемые в лабораториях и на химических предприятиях для контроля химико-технологических процессов.
1.3.1 Спектрометрические методы анализа
Среди перечисленных групп (см. рис.2) обширной по числу методов является группа спектрометрических методов анализа. В отдельных литературных источниках, авторы в зависимости от решаемых задач, спектрометрические методы называют оптическими, либо фотометрическими. Ошибки в этом нет, так как в любом случае используется свойство атомов и молекул определяемого вещества поглощать, отражать или рассеивать электромагнитное излучение, которое регистрируется приборами
Хроматографические
Электрохимические
Спектрометрические



Фотометрический
Электрографиметрический
Газовый


Люминисцентный
Жидкостной
Потенциометрический
Газово-жидкостной
Нефелометрический
Кондуктометрический
Турбидиметрический
Ионообменный
Полярографический

Рефрактометрический
Бумажный
Вольтамперметрический
Поляриметрический
Тонкослойный
Диэлькометрический
Термокондуктометрический
Радиоизотопный
Рис 2. Схема классификации физических и физико - химических методов анализа
Спектрометрические методы предоставляют широкие возможности для получения аналитических сигналов в различных областях спектра электромагнитного излучения – это γ–лучи, рентгеновское излучение, ультрафиолетовое (УФ), видимое и инфракрасное излучение, а также микроволновые и радиоволновые области спектра. Энергия квантов, перечисленных видов излучения, охватывает очень широкий диапазон энергии от 108 до 10-6 эВ, соответствующий диапазону частот от 1020 до 106 Гц.
Природа взаимодействия столь различающихся по энергии квантов с веществом принципиально разная, этим объясняется большое число разнообразных спектрометрических методов анализа. Для решения разнообразных аналитических задач наибольшее значение имеют спектральные методы анализа, оперирующие с излучением видимого, ИК и УФ диапазонов. Эта группа относится к оптическим (фотометрическим) методам анализа и включает:
спектро – фотометрический и фотоколориметрический методы, нефелометрический метод;
абсорбционно – оптический метод;
люминесцентный метод;
поляризационно – оптический метод;
рефрактометрический метод.
В оптических (фотометрических) методах анализа используется связь между составом системы и ее оптическими свойствами: светопоглощением; светорассеянием; преломлением света; вращением плоскости поляризации плоско поляризованного света; вторичным свечением вещества и т.д.
Спектрофотометрический и фотоколориметрический анализы основаны на способности окрашенных растворов, поглощать ультрафиолетовый, видимый или инфракрасный свет. Степень поглощения излучения зависит от концентрации вещества в растворе (абсорбционная спектроскопия).
Нефелометрия основана на способности мутных растворов (содержащих взвесь – меловой раствор, дым и др.) суспензий рассеивать падающий на них пучок света. Интенсивность света рассеянного частицами зависит от концентрации и фиксируется фотоэлементами.
Люминесцентный метод анализа основан на способности свойства веществ, излучать свет под воздействием различных возбуждающих факторов, установлении зависимости этого излучения от концентрации вещества.
Рефрактометрический метод анализа основан на использовании явления преломления света на границе двух сред, на измерении показателя преломления или разницы показателей преломления веществ.
Поляриметрический метод анализа основан на определении содержания вещества по вращению плоскости поляризации. Метод применим только для оптически активных веществ, т.е. способных вращать плоскость поляризации света.
1.3.2 Электрохимические методы анализа
Электрохимические методы анализа: основаны на использовании электрохимических процессов между составом системы и ее электрохимическими характеристиками электропроводностью; электродным потенциалом; поляризацией; количеством электричества и т.д. Для протекания электрохимических процессов используются электролитические ячейки, представляющие собой систему, состоящую из электролитов и электродов, контактирующих между собой. На границе раздела фаз электрод – электролит протекает электрохимическая реакция, в результате которой образуется электродный потенциал.
Электрохимические методы анализа классифицируются в зависимости от процессов происходящих на электродах:
1) методы, не связанные с электродной реакцией, измеряемый сигнал в них является откликом на изменения электрохимических свойств в объёме раствора ( низко- и высокочастотная кондуктометрия );
2) методы, основанные на электродной реакции, в результате которой ток через границу раздела фаз не протекает и на границе раздела фаз устанавливается равновесный потенциал, величина которого зависит от концентрации компонентов, участвующих в электродной реакции (потенциометрия).
3) методы, основанные на электродной реакции между электродом и приэлектродной частью раствора, в ходе которой электроны или ионы переходят через границу раздела фаз, обуславливая возникновение тока (вольтамперметрия, амперметрия, кулонометрия, электрографиметрия).
Широкий круг задач, решаемых с помощью электрохимических методов анализа, делает их конкурентоспособными по отношению к другим инструментальным методам, а в ряде случаев единственно возможными. Методы характеризуются:
высокой чувствительностью (10-3 – 10-7 массовых долей определяемого компонента) - полярография, кулонометрия;
широким интервалом определяемых концентраций (1 – 10-9 моль/л), избирательностью и экспрессивностью – ионометрия и ионографиметрия;
относительной простотой проведения анализа и невысокой стоимостью аппаратуры – кондуктометрия и потенциометрия;
возможностью концентрирования в рамках самого метода (инверсионная вольтамперметрия) или сочетания с другими методами (например, хроматографией, экстракцией);
лёгкостью автоматизации всего аналитического цикла – все методы.
1.3.3 Хроматографические методы анализа
Хроматографические методы анализа (хроматография) предназначены для определения качественного и количественного состава газообразных и жидких веществ. Они широко применяются в научных целях для изучения физико-химических свойств газов и растворов, а в промышленной и лабораторной практике для анализа смеси газообразных, жидких и твёрдых веществ.
Методы основаны на разделении исследуемой смеси веществ между двумя несмешивающимися фазами - подвижной и неподвижной. Подвижная фаза представляет собой поток газа или жидкости, которая непрерывно перемещается вокруг неподвижной фазы (сорбента) – жидкости или твёрдого тела. В результате перемещения подвижной фазы исследуемая смесь разделяется на компоненты за счёт различной поглощаемости (сорбируемости) её составных частей при движении по слою сорбента.
В зависимости от признаков классификации различаются следующие виды хроматографии:
I. По агрегатному состоянию применяемой подвижной фазы: - жидкостная, газовая;
2. По состоянию неподвижной фазы газовой хроматографии - газотвердая, газожидкостная;
3 . По механизму разделения: ионообменная; адсорбционная; распределительная; осадочная;
4. По способу проведения процесса или аппаратному оформлению: колоночная; капиллярная; плоскостная.
Многие физико-химические методы анализа отличаются скоростью проведения определений вследствие высокой их селективности. Чувствительность физико-химических методов анализа превосходит чувствительность графиметрического и титрометрического. Так, чувствительность спектрофотометрических определений составляет 10-3-10-4 , люминесцентного - 10-5-10-6 , полярографического метода анализа – 10-3-10-7 массовых долей (% ) определяемого компонента.
Чтобы получить надежные результаты при использовании физико-химических методов анализа и наиболее полно использовать возможности этих методов, необходимо понимать как процессы химического взаимодействия, так и закономерности возникновения и измерения физических сигналов. Каждая стадия анализа, каждая его операция может быть источником случайных ошибок. Поэтому очень важно уметь оценить с помощью методов математической статистики достоверность полученных результатов анализа.
Физико-химические методы анализа широко используются в практике аналитического контроля протекания химико-технологических процессов на предприятиях, в ходе анализа веществ в производственных и научных лабораториях, а также лабораториях по контролю качества и сертификации продукции.
Особенности физико - химических методов аналитического контроля
Первая особенность заключается в высокой скорости получения результата с помощью физических и физико-химических методов анализа. Скорость анализа на многих производствах имеет большое значение, так как позволяет корректировать технологические процессы, снижать энергетические и др. затраты. На особо опасных производствах, в гражданской обороне в военном деле скорость получения информации о выбросе (появлении или применении) токсичных веществ в воздушное пространство позволяет предотвратить появление неоправданных жертв.
Современные приборы, работающие на принципах физических и физико-химических методов анализа, позволяют получать результаты, как на месте контроля, так и через несколько минут после поступления пробы в лабораторию.
Вторая особенность физических и физико-химических методов анализа не связана с непосредственным определением качества продукции, но благодаря ей представляется возможностью проведения анализа веществ на расстоянии. Примерами таких анализов могут служить:
анализ лунного грунта, выполненный рентгенофлуоресцентным устройством, установленным на луноходе;
определение состава атмосферы, окружающей планету Венера;
исследования атмосферы и грунта на Марсе, которые в настоящее время проводят специалисты США и Евросоюза с использованием методик и средст, разработанных российскими учёными. Разновидностью такого анализа является дистанционный контроль объектов нашей планеты с высокой радиоактивностью или токсичностью, а также на больших глубинах. Такие анализы находят все большее применение для контроля экологической обстановки в промышленно нагруженных районах, особенно при наличии в них ядерных и химических производств.
Третья особенность физических и физико-химических методов анализа позволяет автоматизировать процесс контроля химико-технологических и других производств. Используемые оборудование и приборы работают автоматически и на основании данных анализа регулируют подачу компонентов, поддерживая определенную среду (рН-, концентрацию) в технологическом процессе. Например, при производстве NН>4>NОз автоматические датчики дозируют подачу NН>3 >и НNОз на основании автоматического анализа среды в реакторе - нейтрализаторе (NH>3> + НNОз = NH>4>NO>3>+ Q).
В настоящее время широко применяются автоматические газоанализаторы для контроля воздушной среды, воздуха в шахтах и колодцах, а также для определения мест утечки газов из трубопроводов или ёмкостей и решения других задач.
Четвертая особенность физических и физико-химических методов анализа заключается в возможности исследования веществ без отбора пробы из анализируемого образца, т. е. без его разрушения (недеструктивный анализ). Такие виды анализа проводятся в археологии, медицине, криминалистике и т.д. Иногда такой анализ проводится в какой-то определенной точке образца - локально. Локальный анализ выполняется чаще рентгеноспектральным методом, и широко применим в археологии, криминалистике, минералогии и др. Для целей локального анализа успешно применяется техника лазерной микроспектроскопии.
Пятая особенность физических и физико-химических методов анализа определяется возможностью работать с малыми количествами и концентрациями анализируемых (контролируемых) веществ, составляющих в образце менее 10-3%. Применение в этих случаях классических методов анализа невозможно.
Многие приборы, применяемые в физических и физико-химических методах анализа, совмещены с компьютерами, с помощью которых осуществляется управление химико-технологическими процессами, проводятся расчеты, статистическая обработка полученных данных и решаются другие аналитические задачи.
1.5 Выбор метода анализа
Выбор более рационального и точного метода лабораторного анализа вещества зависит от многих факторов и представляет довольно трудную задачу, так как обычно связан с необходимостью решения многовариантных задач. Поэтому для его проведения привлекаются специалисты высокой квалификации, знающие методики и особенности проведения анализа, а также умеющие пользоваться соответствующим оборудованием.
Аналитический контроль производимого вещества в ходе протекания автоматизированных химико-технологических процессов, как правило, одновариантен для точки контроля, которых может быть достаточно много. Он осуществляется в соответствии с заранее отработанной и, как правило, отлаженной программой выпуска продукции (технологией). Вместе с тем, изменение химического состава перерабатываемых веществ и образование новых веществ, отвечающих заданным требованиям, обязывает операторов постоянно контролировать режимы процессов. При этом измеряются параметры, как промежуточных продуктов, так и соответствие выпускаемой продукции заданным требованиям, что позволяет судить о её качестве.
В лабораторных условиях наиболее просто решить задачу об определении количественного содержания одного элемента (вещества) в анализируемом продукте. Если определяемый элемент является основным компонентом анализируемого объекта и его содержание велико, применяются химические методы анализа - гравиметрический или титриметрический. Если концентрация определяемого элемента очень мала, то анализ проводится с помощью физико-химических методов анализа - оптическим, электрохимическим, хроматографическим или каким либо другим.
Выбор метода зависит также от того, какое количество проб подлежит анализу и с какой частотой.
Единичные анализы или небольшое их количество, как правило, целесообразно проводить химическими методами. Применение инструментальных методов для единичного анализа - нецелесообразно, т.к. много времени займет предварительная калибровка аппаратуры построение градуировочных графиков, стандартных образцов для сравнения и т.д. При необходимости проведения анализа большой серии проб приблизительно одинакового состава применение инструментальных методов не только оправдано, а просто необходимо.
Например, большое количество кальция в исследуемом образце определяется гравиметрическим или титриметрическим методом, относящимся к химическим методам. Причем, если нужна высокая точность, а длительность анализа не регламентируется – применяется гравиметрический (весовой) метод анализа. Если не требуется высокой точности, но нужен срочно результат - применяется титриметрический (объемный) метод анализа, в этом случае можно быстро оттитровать кальций комплексонометрически - это быстро, хотя точность анализа ниже.
Очень малые содержания кальция в большой серии однотипных проб определяется инструментальными методами анализа, однако выбор метода будет зависеть от наличия соответствующей аппаратуры. Выбор метода осложняется, если анализируемое вещество содержит много сопутствующих компонентов в различных количественных соотношениях, так как приходится учитывать их химическую природу.
Микроколичества цинка легко определяются полярографически, но большие количества меди и кадмия мешают этому определению, т.к. они восстанавливаются раньше цинка, поэтому медь и кадмий нужно предварительно удалить. Для проведения таких операций нужно знать свойства определяемых катионов. Если нужно провести такой анализ – применяется экстракционно-фотометрический метод с дитизоном, который образует с цинком окрашенный комплекс, экстрагируемый тетрахлоридом углерода. Этот прием проводится в присутствии насыщенного раствора тиосульфата, который образует тиосульфатные комплексы с медью и кадмием, не способные экстрагироваться тетрахлоридом углерода, а потому остаются в водном растворе. Окрашенный экстракт соединения цинка с дитизоном в среде тетрахлорида углерода - фотометрируется.
Железо в растворе можно легко анализируется, применением гравиметрического метода, при этом в качестве осадителя используется гидрат аммиака. Однако этот метод нельзя применять в присутствии титана, который тоже образует нерастворимый гидроксид.В этом случае целесообразнее применить 8-оксихинолин, который полностью осаждает железо уже при рН=3, в то время как титан остается в растворе.
Одним из ответственных моментов в титриметрическом методе является фиксация эквивалентной точки. Фиксация проводится, обычно, по изменению окраски индикатора.
Иногда применение цветных индикаторов оказывается затруднительным или вовсе невозможным, например, при титровании мутных, сильноокрашенных или очень разбавленных растворов. А для некоторых реакций вообще не найдены соответствующие индикаторы. В таких случаях используются физико-химические методы, т.е. в ходе титрования наблюдаются не изменение окраски индикатора, а изменение электрохимических показателей титруемого раствора: электропроводности (кондуктометрическое титрование), окислительно-восстановительного потенциала (потенциометрическое титрование) и т.д. Точка эквивалентности определяется не путем визуального наблюдения за изменением окраски индикатора, а с использованием специального прибора, дающего объективные показания.
Сложная аппаратура и приборы, применяемые в практике контроля качества продукции, требуют высокой эрудиции и знаний в области химии, физики, математики, знаний принципиальных схем работы применяемых приборов, умения правильно применять аппаратуру и приборы для получения объективных данных. Всё это позволит своевременно принять меры для выпуска качественной продукции, оценить её безопасность и возможные изменения при использовании, а также разрешать возникающие споры между производителем и потребителем, и при необходимости защитить интересы одной из сторон.
1.6 Сигнал как информативная функция состава вещества
В основе физико-химических методов анализа лежит измерение соотношений между составом и свойствами исследуемых продуктов. В большинстве случаев эта зависимость очень сложная. Часто одно и то же свойство соответствует различным значениям состава, т.е. является многозначной функцией состава, что затрудняет использование его для аналитических целей. Поэтому в физико-химических методах анализа результаты исследований выражаются в виде диаграммы “состав-свойства” и используются только те участки, где состав определяет свойство.
В ходе физических и физико-химических методах анализа измеряются величины, отражающие физико-химические свойства веществ, такие как электрическая проводимость, поглощение и преломление света и т.д. Каждое изменение регистрируется в виде аналитического сигнала, являющегося информативной функцией состава вещества, которую строят с использованием стандартных образцов. Эти сигналы регистрируются специальными приборами, позволяющими в зависимости от их интенсивности определять количество вещества в исследуемом продукте. Например, в прямой кондуктометрии таким регистрируемым аналитическим сигналом является электропроводность, которая зависит от концентрации растворенного вещества, а в методе прямой потенциометрии - сигналом является потенциал индикаторного электрода, который также зависит от концентрации определяемого вещества. В фотоколориметрии в качестве аналитического сигнала измеряется оптическая плотность серии стандартных растворов, имеющих разные концентрации, в потенциометрии – электродный потенциал и т.д.
Для решения практических задач по определению качества веществ возникает специфическая, ответственная и достаточно сложная задача стандартизации (эталонирования) самих объектов анализа. Сложность задачи объясняется многообразием веществ, отличием их химического состава и физико- химических свойств. Отсутствие эталонов, идентичных анализируемым пробам, как правило, приводит к ошибкам, поэтому в лабораторной практике применяют корректирующие методы.
Под стандартным образцом понимается специально приготовленное вещество, предназначенное для обеспечения правильности химического анализа. К стандартам наиболее высокого класса точности относятся образцы изготовленные централизовано, более низкий класс точности имеют стандартные образцы предприятий и лабораторий.
Химический состав и физико- химические свойства стандартного образца официально аттестованы, и данные о содержании компонентов и области его применения указаны в аттестате. Если стандартный образец не имеет официального статуса, то он называется веществом сравнения. Часто в качестве эталона используются химически чистые вещества, содержащие не более 0,05% примесей.
Число типов стандартных образцов, имеющих официальный статус, ограничено. Острый дефицит эталонов ощущается в таких отраслях, как органический синтез производство, пластмасс, синтетических смол и других отраслях химической промышленности. Крайне необходимы эталоны для целей мониторинга за состоянием окружающей среды. На предприятиях пищевой промышленности также должны быть соответствующие стандарты, но, судя по изменяющейся продукции, поступающей в продажу, они явно не всегда принимаются в качестве эталонов. Вероятно, контроль качества продукции производится статистическими методами, в основе которых лежит объём продажи.
1.7. Основные приемы получения результата физико-химическими методами контроля
Практически во всех физико-химических методах аналитического контроля применяются два основных приема получения результата - прямыми измерениями и косвенными измерениями.
1.7.1 Прямые измерения
При прямых измерениях используется зависимость аналитического сигнала от природы анализируемого вещества и его концентрации. В спектроскопии, например, длина волны спектральной линии, определяет свойство природы вещества, а количественной характеристикой является интенсивность спектральной линии.
Поэтому, при проведении качественного анализа фиксируют сигнал, а при проведении количественного анализа - измеряют интенсивность сигнала.
Между интенсивностью сигнала и концентрацией вещества всегда существует зависимость, которая может быть представлена выражением 1.7.1.
I =K · С, (1.7.1)
где: I — интенсивность аналитического сигнала;
K — константа;
С — концентрация вещества.
В аналитической практике прямые измерения применяются наиболее часто, к ним относятся: метод градуировочного графика; метод молярного свойства; метод добавок.
Метод градуировочного графика
Метод градуировочного графика, применяется в большинстве физико- химических методов анализа. Для его реализации измеряется интенсивность аналитического сигнала у серии стандартных образцов или растворов и строится градуировочный график(рис.3, рис.4) функции 1.7.2.
I=f (C), (1.7.2)
где:I — интенсивность сигнала;
C — концентрация компонента, определяемого в стандартном образце или растворе.
Затем в этих же условиях измеряется интенсивность аналитического сигнала, в анализируемой пробе - I>х> , и по градуировочному графику находится концентрация анализируемого образца – С>х>.
Если градуировочный график описывается уравнением y = b•C, то он может быть построен по одному эталону, а прямая будет выходить из начала координат. В этом случае измеряются аналитические сигналы для одного стандартного образца и пробы. Далее рассчитываются погрешности, и строится корректирующий график.
Если градуировочный график строится по уравнению y = a + b•C, то необходимо использовать как минимум два эталона. Реально для уменьшения погрешности используются от двух до пяти эталонов.
Интервал концентраций на градуировочном графике должен охватывать предполагаемую область анализируемых концентраций, а состав стандартного образца или раствора должен быть близок к составу анализируемого. На практике это условие редко достигается, поэтому желательно иметь широкий набор стандартных образцов разнообразного состава.
В уравнении прямой y = a + b•C величина b характеризует наклон прямой и называется коэффициентом инструментальной чувствительности. Чем больше b , тем больше наклон графика и тем меньше погрешность определения концентрации.
Может применяться и более сложная зависимость, кроме того, перевод функций в логарифмические координаты позволяет ослабить влияние побочных процессов и предотвращает появление ошибки.
Градуировочный график должен строиться непосредственно перед измерениями, однако в аналитических лабораториях при выполнении серийных анализов используют постоянный, заранее полученный график. В этом случае необходимо проводить периодические проверки правильности результатов анализов во времени. Частота контроля зависит от величины серии проб. Так, для серии из 100 проб выполняют один контрольный анализ на каждые 15 проб.
Метод добавок
Когда состав пробы неизвестен или о нём имеется недостаточно данных, а также когда отсутствуют адекватные стандартные образцы, применяется метод добавок. Он позволяет в значительной степени устранить систематические погрешности, когда существует несоответствие между составом эталонов и проб.
Метод добавок основан на введении в серию одинаковых по массе и объёму проб анализируемого раствора (А>х>) точно известного количества определяемого компонента (а) с известной концентрацией (С>а>). При этом измеряется интенсивность аналитического сигнала пробы до введения ( I>x> ) и после введения дополнительного компонента (I>х+а>). Концентрация вещества (С>х>) в исследуемом растворе рассчитывается по выражению 1.7.3 или находится графически.
А>х>/А>х+а >= Сх/А>х>+С>а>
или С>х> = С>а> А>х> / А>х+а >– А>х >
Число проб с добавками переменных количеств определяемого компонента может варьироваться в широких пределах.
Метод молярного свойства
В этом методе измеряется интенсивность аналитического сигнала нескольких стандартных образцов или растворов и рассчитывается среднее молярное свойство по выражению 1.7.4.
Ā=1/n>i>∑I/С , (1.7.4)
где: Ā – среднее молярное свойство;
n>i> – количество измерений i-х стандартных образцов;
I – интенсивность сигнала;
С – концентрация
Для определения концентрации анализируемого компонента измеряется интенсивность сигнала у анализируемой пробы, а расчет проводится с использованием выражения 1.7.5.
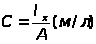 (1.7.5)
(1.7.5)
Метод предполагает строгое соблюдение соотношения
I = А · С в области
анализируемых концентраций.
1.7.2 Косвенные измерения
Косвенные измерения применяются при титровании анализируемой пробы кондуктометрическим, потенциометрическим и некоторыми другими методами.
В этих методах в процессе титрования измеряется интенсивность аналитического сигнала - I и строится кривая титрования в координатах I - V, где V - объем добавляемого титранта в мл.
По кривой титрования находится точка эквивалентности и проводится расчет, по соответствующим аналитическим выражениям 1.7.6.
Q
в-ва = Т г/мл ·
Vмл(экв)
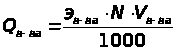 (1.7.6)
(1.7.6)
Виды кривых титрования весьма многообразны, они зависят от метода титрования (кондуктометрическое, потенциометрическое, фотометрическое и т.д.), а также от интенсивности аналитического сигнала, зависящего от отдельных влияющих факторов.
2. Автоматизация аналитического контроля продукции химико-технологических производств
Автоматизированный аналитический контроль является обязательным элементом управления химико-технологическими процессами автоматизированных производств различной продукции. Он выполняется проведением определённой совокупности операций, контролирующих протекание технологических процессов непрерывного получения продукции заданного качества
Контроль представляет собой процесс измерения параметров продукции, объединённый с принятием решения на применение предупреждающих и корректирующих действий, рис. 2.1.,
Технологическая
система производства
Управляющий
орган


Прогноз
качества
продукции
Корректирующие
действия
Предупреждающие
действия



Рис 2.1. Место автоматизированного аналитического контроля в технологической системе производства продукции
Целью проведения контроля является оперативное получение объективной информации о состоянии технологической среды в различных точках технологической системы производства. Оперативность достигается использованием автоматических анализаторов (аналитических приборов), позволяющих измерять параметры технологической среды, которые характеризуют её элементарный, молекулярный или фазовый состав.
Автоматические анализаторы являются устройствами, измеряющими конкретный (заданный) физический параметр выбранного компонента технологической среды. По изменению параметра оценивается состояние протекающего химико-технологического процесса, прогнозируется качество выпускаемой продукции, и производятся (автоматически или операторами) соответствующие корректирующие и предупреждающие действия.
2.1 Измеряемые параметры и их использование для анализа качества продукции
Под параметрами продукции понимаются показатели, характеризующие состав контролируемой технологической среды и свойства, входящих в неё веществ.
Состав технологической среды зависит от количества, входящих в неё отдельных веществ (компонентов) и может быть выражен числом молей или массой компонентов в граммах или других единицах массы. Однако в практике аналитического контроля состав выражается через концентрацию компонентов (С = м / М), которая учитывает взаимосвязь между массой отдельного компонента в пробе (м) и общей массой пробы (М). Наиболее распространёнными единицами измерения концентрации являются:
для жидкостей-мг/см3, г/см3, % по массе или объёму;
для газов-мг/м3, г/м3, % по объёму.
Свойства веществ характеризуются численными значениями физических или физико-химических величин (плотности, вязкости, электропроводности и др.), которые могут быть использованы для измерения.
Практическое выполнение аналитических измерений основано на использовании взаимосвязи между составом анализируемого вещества (концентрациями его компонен тов) и величинами, характеризующими его физические и физико-химические параметры, выражение 2.1.
y = f ( С>1>, С>2>,…,С>i>,…,С>п> ), (2.1)
где: y-измеряемый параметр анализируемого вещества;
С-концентрация компонентов;
п -общее число компонентов в контролируемой продукции.
2.2 Технологическая среда химико-технологических процессов и её свойства
К технологической среде химико-технологических процессов относятся, находящиеся в технологических аппаратах, продукты (В отдельных литературных источниках технологическую среду называют объектами аналитического контроля и обозначают ОАК). Они отличаются составом и свойствами перерабатываемых и производимых веществ, которые могут представлять собой гомогенную или гетерогенную среду, состоящую из нескольких фаз (как правило, от одной до трёх). В гомогенной среде измеряется содержание одного или нескольких компонентов, а в гетерогенной среде – содержание одного или нескольких компонентов в одной из фаз.
В соответствии с определяемым компонентом выбирается измеряемый физический параметр контролируемой технологической среды, его выбор зависит от двух факторов:
наличия соответствующего оборудования, которое может быть применено для обеспечения аналитического контроля производства;
наличия достоверных данных о физических свойствах контролируемой и анализируемой технологической среды. Например, для измерения концентрации водорода в газовых смесях используются термокондуктометры, так как водород обладает значительно большей теплопроводностью, чем другие газы. Для измерения концентрации кислорода в газовых смесях применяют термомагнитные газоанализаторы, так как молекулы кислорода обладают ярко выраженными парамагнитными свойствами.
Подлежащая аналитическому контролю технологическая среда, как правило, представляет собой жидкости, газы, суспензии, эмульсии, дымы, туманы или их смеси. При проведении контроля они в большинстве случаев подвергаются - фильтрации, нагреванию или охлаждению и другим преобразованиям. Это происходит при движении технологической среды в технической системе, представляющей собой транспортную коммуникацию от точки отбора пробы до места установки датчиков контроля. Тем самым обеспечивается перевод её (анализируемой среды) в состояние, удобное для контроля с помощью анализаторов.
В ходе контроля из-за повышения температуры, давления или роста концентрации веществ в технологических процессах могут происходить изменения в фазовом состоянии технологической среды. Подобное состояние должно прогнозироваться при разработке технологий контроля, так как результаты анализа будут необъективными.
В производственной практике наиболее встречающимися технологическими средами являются, рис.2.2:
однофазная газовая среда;
газовая среда, содержащая неустойчивые аэрозоли;
газовая среда, содержащая неустойчивые и устойчивые (устойчивые и неустойчивые) аэрозоли;
однофазная (чистая) жидкая среда;
суспензии (жидкая среда, содержащая твёрдые частицы);
эмульсии (жидкая среда, содержащая частицы органических или элементоорганических веществ).
Технологическая среда


Газовая
Жидкостная




Чистая
Однофазная
Суспензии
Содержащая аэрозоли
Эмульсии





Рис. 2.2. Классификация фазовых состояний технологической среды
Однофазная газовая среда характеризуется отсутствием аэрозоля и не изменяет агрегатного состояния при изменении температуры и давления при проведении аналитического контроля.
Газовая среда с неустойчивыми или устойчивыми и неустойчивыми аэрозолями остаётся однофазной при изменении температуры и давления в пределах, требуемых для проведения анализа. Например, такими средами являются газы, обезвоживаемые твёрдыми или труднолетучими осушителями (серной кислотой).
Газовая среда, содержащая устойчивые и неустойчивые аэрозоли (пар, туман, дымы) при изменении температуры и давления изменяет своё агрегатное состояние и частично конденсируется, Например, автоматизированная система контроля паров на различных уровнях ректификационной колонки отличается многозвенностью подсистем, обеспечивающих выделение мешающих компонентов из смеси механическими или физико-химическими методами.
Чистая жидкая среда не содержит диспергированных частиц (жидких, твёрдых, газообразных). При транспортировке через автоматизированную систему её агрегатное состояние и свойства не изменяются. Например, к такому типу контролируемых жидкостей относится обессоленная вода (очищенная от солей жёсткости) используемая в котлах котельных для получения водяного пара.
К суспензиям относится жидкая среда, содержащая твёрдые частицы. Жидкая фаза суспензии не изменяет своего агрегатного состояния при изменении температуры и давления в заданных пределах.
Различаются суспензии с неустойчивой или сочетающей в себе неустойчивую и устойчивую твёрдые фазы. Твёрдые частицы неустойчивой фазы осаждаются под действием гравитационных сил или фильтруются, например, производство оксохлорида меди в результате взаимодействия водно-меловой суспензии с раствором хлорида меди. Примером суспензий с неустойчивой и устойчивой твёрдой фазой служат сконденсированные продукты высокотемпературного органического синтеза, в которых содержатся твёрдые частицы углерода (сажи). Они образуются в результате частичного разложения реагентов при высокой температуре, и для их удаления требуется сверхтонкая фильтрация производимого продукта.
К эмульсиям относится жидкая среда, содержащая как крупные, так и мелкие частицы диспергированных органических или элементоорганических веществ. Примерами эмульсий являются продукты полимеризации в растворах органических и элементоорганических вещ(полистирол, поливинилхлорид и др.).
Анализируемой средой эмульсий является как сплошная, так и дисперсная фаза, которые могут изменять агрегатное состояние при изменении температуры и давления. Кроме того, дисперсная фаза жидкой среды при транспортировке её в автоматизированной системе подготовки к анализу может коагулироваться. Поэтому для анализа дисперсные фазы разделяются и специально готовятся (термически обрабатываются и дозируются).
Жидкая среда может содержать растворённый газ, концентрация которого изменяется при изменении температуры и давления, а концентрация твёрдой и жидкой фазы остаётся постоянной. Примерами таких сред являются продукты хлорирования водно-органических суспензий. Сплошной фазой в них является водный раствор, в котором с высокой точностью поддерживается заданное значение рН.
В технологической среде, представляющей собой насыщенный раствор, даже при незначительном изменении температуры могут образовываться диспергированные частицы. Для проведения анализа такая среда подвергается фильтрации, термической обработке, при необходимости разбавлению, дозировке.
2.3 Автоматизированные системы аналитического контроля
Автоматизированные системы аналитического контроля продукции обычно монтируются как в специально оборудованных производственных помещениях (анализаторных), так и в непосредственной близости от аппаратов, в которых протекают технологические процессы производства продукции. Они представляют собой совокупность, взаимодействующих между собой технологической среды и технических устройств отбора, подготовки и анализа пробы, а также обработки и отображения полученных данных.
Взаимодействие между составляющими автоматизированной системы осуществляется в соответствии с разработанной методикой.
2.3.1 Методика автоматизированного аналитического контроля
Методика автоматизированного контроля химико-технологических процессов, как правило, разрабатывается на стадии проектирования системы контроля конкретного производства. При её разработке применяются типовые методы анализа веществ, а в отдельных случаях разрабатываются новые. Практика организации аналитического контроля не исключает возможности применения методик, используемых на предприятиях, выпускающих одинаковую или близкую по составу продукцию. Однако методики сторонних предприятий должны быть адаптированы к условиям конкретного производства.
Как документ методика оформляется в виде пояснительной записки к карте аналитического контроля. В ней подробно описываются порядок отбора пробы, условия её транспортирования, подготовки, измерения и отображения параметров контролируемой среды, а также организация сброса проконтролированного продукта в технологическую систему. Перечисляются операции, подлежащие выполнению с указанием используемого оборудования и химических реактивов, а также приводится математический аппарат расчета прогнозируемых систематических и случайных погрешностей.
Для обеспечения качества и единства полученных результатов анализа в методике обосновывается периодичность поверки автоматизированной системы аналитического контроля. Кроме того, излагаются функции ведомственной метрологической службы, как во время поверки, так и в межповерочный период.
Особое внимание в методике уделяется параметрам объектов аналитического контроля:
составу и свойствам контролируемых компонентов или технологической среды в целом;
мешающим компонентам, изменению концентрации и фазового состояния технологической среды;
внутренним и внешним факторам, оказывающим влияние на процесс контроля.
С точки зрения энергетического подхода проба может характеризоваться двумя группами параметров:
1) внутренними - определяемыми физическими параметрами, которые функционально связаны с движением молекул ,атомов, ионов, электронов, ядер, функциональных и молекулярных групп, а в случае неустановившегося процесса, временем;
2) внешним – измеряемыми физическими параметрами, зависящими от расположения внешних (по отношению к пробе) тел и характеризующими параметры пространства, времени, силовых полей, излучения.
Внутренние и внешние параметры пробы связаны между собой, однако определяемые параметры непосредственно связаны только с составом пробы.
Можно отметить, что преобразования и измерения параметров пробы связаны с воздействием на неё различных полей или веществ. В зависимости от характера данного воздействия различаются следующие преобразования пробы:
1. Химические. Если воздействие на пробу приводит к изменению состава системы пробы – источник воздействия;
2. Физико-химические. Если воздействие на пробу изменяет состав системы пробы – источник воздействия, а также вызывает пространственное или пространственно-временное разделение 4компонентов пробы;
3. Физические. Если воздействие на пробу приводит к изменению её свойств при неизменности состава;
4. Комбинированные, состоящие из различных вариантов рассмотренных воздействий.
Любые изменения в процессах (увеличение или уменьшение числа фаз, разделение их, разбавление среды и др.) в методике оговариваются специально, и каждому из них даётся точная количественная оценка. Поэтому при разработке методики контроля особые требования предъявляются к реализуемым методам измерения параметров веществ. Они во всём интервале изменения концентрации определяемого компонента должны обладать максимально возможной параметрической чувствительностью (ПЧ) и наиболее полно соответствовать зависимости 2.2.
n
ПЧ = П с ∕ ∑ П с → max, (2.2)
K=1
где: -интервал изменения концентрации -го (определяемого) и -го (неопределяемого) компонента;
-значение выбранного параметра, относительно которого измеряется соответственно концентрация -го и -го компонента многокомпонентной смеси.
2.3.2Составные части автоматизированной системы аналитического контроля
Аналитический измерительный процесс (рис.2.3) в автоматизированных системах контроля условно может быть разделён на четыре этапа:
отбора пробы на анализ;
транспортирование пробы к анализатору;
подготовку пробы к анализу;
непосредственное проведение аналитических измерений и обработки измерительной информации.
На этапе отбора пробы для анализа наиважнейшее значение придаётся месту монтажа пробоотборного устройства в технологической системе. Оно должно обеспечивать:
постоянство пропускной способности отбираемой пробы технологической среды;
соответствие пробы основной массе контролируемой технологической среды по физико-химическим свойствам.
От выбора места монтажа на технологической линии пробоотборного устройства зависят условия отбора пробы на анализ. Они оказывают влияние на стабильность функционирования узлов подготовки пробы к анализу и работу анализатора и, как следствие, повлияет на достоверность результатов измерений параметров пробы.
Этапы транспортирования и подготовки могут быть разделены только условно.
Подготовка пробы к анализу начинается в транспортной коммуникации от пробоотборного устройства до места установки датчика для измерения параметров. Она состоит обычно из ряда элементов, каждый из которых обеспечивает определённый вид преобразования технологической среды - фильтрацию, нагревание или охлаждение, разделение фаз и др. Таким образом, проба переводится в состояние обеспечивающее проведение контроля с помощью автоматического анализатора.
Как показывает опыт, обычно контролируемая технологическая среда подвергается таким преобразованиям, в результате которых среда анализируемой пробы становится
В
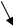

 нешние
влияющие факторы
нешние
влияющие факторы

х
Отбор пробы на анализ

Подготовка
к анализу


Проведение
аналитических измерений
у


Анализатор
Индикатор
Усилитель сигналов
Микропроцессор
Преобразователь
Параметры промежуточных точек контроля
Внутренние влияющие факторы







Сброс (утилизация)

Рис. 2.3. Операционная схема автоматизированного аналитического контроля
гомогенной и соответствует требованиям, определённым документацией применяемого анализатора.
При транспортировании и подготовке пробы к анализу продукт может изменить фазовое состояние из-за изменения температуры. Состав пробы может измениться вследствие сорбции или хемосорбции на внутренних поверхностях транспортных коммуникаций, а также продолжающихся химических превращений и других процессов. Величина вносимой погрешности на этом этапе, может оказаться значительной и многократно превосходить погрешность анализа, нормированную для приборов. Такие погрешности не всегда удаётся предотвратить, однако их можно учесть, если они остаются неизменными во времени (систематическими).
Пробы некоторых технологических сред не подвергаются преобразованию, и аналитический контроль проводится без дополнительной подготовки. В таких случаях аналитический датчик монтируется непосредственно в технологическом аппарате или технологическом трубопроводе. Для обеспечения надёжного контроля в таких условиях должна быть гарантирована стабильная работоспособность чувствительного элемента анализатора.
Автоматизированная подготовка однофазной газовой среды наиболее простая и заключается в фильтрации её для удаления продуктов эрозии технологических трубопроводов и термостатирования. В отдельных случаях узлы подготовки могут вообще отсутствовать.
Автоматизированная подготовка газовой среды с неустойчивыми или устойчивыми и неустойчивыми аэрозолями к контролю потребует проведения предварительной очистки от аэрозолей, термической обработки и стабилизации расхода.
Чистая жидкая среда не содержит диспергированных частиц (жидких, твёрдых, газообразных) и не требует специальной подготовки для проведения анализа.
При подготовке суспензий к анализу подлежат удалению разложившиеся реагенты проведением сверхтонкой
фильтрацией пробы технологической среды.
Для анализа эмульсии её дисперсные фазы разделяются и специально готовятся - подогреваются и дозируются.
Автоматизированная подготовка к анализу жидкой среды содержащей растворённый газ включает фильтрацию водного раствора и стабилизацию расхода выделенного продукта через автоматический анализатор.
Автоматизированная подготовка насыщенного раствора технологической среды к анализу включает выполнение следующих операций: фильтрацию, термическую обработку, разбавление (при необходимости), дозировку.
Непосредственное измерение параметров пробы производится автоматическими анализаторами. В соответствии с определяемым компонентом выбирается измеряемый физический параметр технологической среды, изменение которого должно наиболее полно характеризовать изменение концентрации определяемого компонента в этой среде. Регистрирует изменение физического параметра чувствительный элемент анализатора. Получение достоверных результатов анализа и снижение погрешностей достигается обеспечением нормального функционирования датчика, что является прямой обязанностью обслуживающего персонала.
2.3.3 Чувствительный элемент датчика автоматического анализатора
Чувствительный элемент является составной частью датчика применяемого анализатора и наиболее важной частью автоматизированной системы контроля технологической среды. Он представляет собой устройство, способное генерировать информацию, полученную от его физического или физико-химического взаимодействия с технологической средой.
К основным требованиям, предъявляемым к чувствительному элементу, относятся:
высокая параметрическая чувствительность к изменению концентрации определяемого компонента;
избирательность, быстродействие, стабильность работоспособности и коррозионная стойкость к анализируемой среде;
доступность и простота регенерации;
надёжность в работе;
удобство обслуживания.
Взаимодействие чувствительного элемента с анализируемой средой может осуществляться прямым контактом или через перегородки.
Прямой контакт чувствительного элемента с технологической средой используется в электрохимических, тепловых, радиоизотопных, газодинамических и других анализаторах. Датчик этих анализаторов монтируется непосредственно в местах автоматизированного контроля среды, которая воздействует непосредственно на чувствительный элемент.
Перегородки применяются для пропускания только того материального потока анализируемой технологической среды, который должен оказывать воздействие на чувствительный элемент. Например, к таким потоком относятся:
поток воздуха эквивалентный давлению анализируемой среды на эластичную мембрану в анализаторах плотности;
световой поток через оптически прозрачные перегородки в оптико- акустических анализаторах;
тепловой поток через защитную стеклянную оболочку в термокондуктометрических анализаторах химически агрессивных сред.
Могут применяться перегородки при контроле технологической среды, включающей несколько фаз. В этом случае возникает необходимость выделения фазы из потока технологической среды, которая характеризует её параметры.
Независимо от того, как будет взаимодействовать чувствительный элемент с анализируемой средой, её физическое состояние должно оставаться постоянным. Однако для предотвращения появления нарушений в его параметрической чувствительности необходимо исключить влияние физических параметров окружающей среды – температуры, давления, влажности воздуха, магнитных полей, производственных вибраций, статического электричества, шагового напряжения и др. Важнейшим условием полноценного его функционирования является поддержание в должном техническом состоянии коммуникаций с измерительным преобразователем и другой аппаратурой.
Обеспечение длительной эксплуатации чувствительного элемента в датчике анализатора и получение объективной информации о состоянии контролируемой технологической среды достигается обеспечением выполнения следующих требований:
1. Чувствительный элемент должен взаимодействовать только с представительной частью анализируемой среды;
2. Недопустима установка чувствительного элемента в застойной зоне контролируемой технологической среды;
3. Режим обтекания чувствительного элемента анализируемой средой, а также её температура и давление должны находиться в пределах определённых методикой контроля.
4. Поверхность контакта чувствительного элемента должна всегда оставаться чистой и неизменной во времени.
В зависимости от решаемых задач и структуры автоматизированной системы информация от чувствительного элемента через датчик передаётся на расстояние по специальным коммуникациям связи к приборам, где обрабатывается, при необходимости усиливается и отображается на индикаторе.
2.3.4. Структуры приборов автоматизированного аналитического контроля
В зависимости от исполнения аналитические приборы могут изготавливаться в виде единой конструкции и в виде комплекта, состоящего из различных блоков, каждый из которых выполняет определённую функцию: обработки, измерения, нормирования усиления и отображения измеренных величин.
Общий принцип работы аналитического прибора заключается в следующем Первым в измерительной цепи аналитического прибора размещается первичный измерительный преобразователь. К нему подведён определяемый физический параметр, зарегистрированный чувствительным элементом датчика. Физический параметр в первичном измерительном приборе преобразуется в выходной электрический сигнал. В последующих блоках сигнал соответствующим образом преобразуется (усиливается, нормируется, видоизменяется и т. д.) в удобную для контроля форму. Контролируется сигнал посредством измерения его величины с помощью применения электронных автоматических мостов и потенциометров.
Потребности практики химико- технологических производств удовлетворяются изготовлением жёстких и гибких структур приборов для автоматизированного аналитического контроля, рис.2.4.
Структуры аналитических приборов
Жесткие
Одноканальная
Двухканальная
Компенсационная
Гибкие
С использованием микропроцессоров
С использованием ЭВМ








Рис. 2.4. Классификация структур автоматизированных систем
К жёстким системам относятся следующие типы структур: одноканальная, дифференциальная (двухканальная), компенсационная.
Одноканальная структура обеспечивает непосредственный отсчёт параметров состава или свойств анализируемого вещества. В ней последовательно располагаются основные и вспомогательные элементы, участвующие в процессе контроля технологической среды, рис 2.5.
1
2
3
x
y
y>2>
Рис. 2.5. Схема одноканальной структуры x-входные параметры (состав или свойства);
1-первичный измерительный преобразователь (ПИП);
y-выходной сигнал удобный для дальнейшего преобразования в системе;
2.-нормирующий преобразователь;
3.-вторичный прибор;
y>1>, y>2>-преобразованные во втором и третьем приборах сигналы.
Основным недостатком одноканальной структуры является отсутсвие управляющего воздействия. Это приводит к нарушению гибкости структуры по информативному каналу, что снижает её метрологические и эксплуатационные свойства .
Одноканальная структура (непосредственного отсчёта)исторически рассматривается как прообраз других структур.
Структура двухканальная дифференциального типа включает рабочий и сравнительный каналы, рис. 2.6.
Основное достоинство структур дифференциального типа состоит в том, что второй (сравнительный) канал позволяет повысить информационный уровень первого (рабочего) канала и снизить влияние помех на процесс контроля. Метрологические характеристики двухканальной структуры выше, чем у структуры непосредственного отсчёта.
Рабочий канал
1
2
x
y
y>1>
Сравнительный канал
11
21
x0
y0
y>1>0
3
4



y>2>
y>3>
Рис. 2.6. Схема двухканальной структуры дифференциального типа
1, 11 –первичный измерительный прибор;
2, 21 –нормирующий преобразователь;
3 –блок сравнения
4 –вторичный прибор.
Основными недостатками структуры являются: низкая скорость анализа и отсутствие управляющих воздействий , что снижает гибкость метода контроля.
Управляющие воздействия на процесс анализа реализуются в структурах аналитических приборов компенсационного типа, рис. 2.7.
В данной структуре реализован принцип компенсации. Он заключается в компенсации неизвестного значения информационного сигнала о составе или свойствах анализируемого вещества известным значением, полученным с помощью специальных средств. В момент компенсации отсчитывается значение информационного сигнала.
Эта структура превосходит по своим показателям предыдущие структуры, но не обеспечивает достаточную гибкость режимов контроля и управления. В целом гибкость определяется наличием следящих операционных систем, чем их больше, тем выше гибкость структуры.
6
7
1
2
x
y
y>1>
11
21
x0
y0
y>1>0
3
4

y>2>
y>3>
5



7

6
Рис. 2.7. Схема структуры компенсационного типа
1, 11 –первичный измерительный прибор;
2, 21 –нормирующий преобразователь;
3 –блок сравнения;
4 –усилитель;
5 –блок управления;
6 –вторичный прибор;
7 –операционная система.
Повышение гибкости достигается за счёт внедрения в структуру аналитического прибора микропроцессорных средств, обеспечивающих автоматизацию процесса определения контролируемого свойства вещества или параметра его состава, а также проведение вычислительных операций.
Гибкая структура (рис.2.8) аналитического прибора позволяет учитывать влияние параметров окружающей среды на точность измерений и получать информацию о составе и свойствах анализируемых веществ в режиме реального времени.
Объект
аналитического контроля
1
2
3






x
x>1>
Рис. 2.8. Обобщённая схема гибкой структуры аналитического прибора
1 –информационный канал;
х -входные параметры, определяющие состав и свойства анализируемых веществ;
2 –корректирующий канал;
х>1 >–входные параметры, которыми могут быть неконтролируемые компоненты анализируемых веществ;
3 –микропроцессорный блок.
Влияние параметров окружающей среды изучается в ходе разработки технологии или производства конкретной продукции и учитывается при создании методики её аналитического контроля. Полученные результаты реализуются в аналитических приборах с гибкой структурой проведения аналитического контроля.
2.3.5 Требования, предъявляемые к приборам аналитического контроля
В основу требований, предъявляемых к приборам, положен принцип, направленный на обеспечение эффективного использования разработанного метода аналитического контроля. Он может быть реализован при условии, если приборы будут отвечать статическим и динамическим критериям эффективности. К основным из них относятся:
точность и чувствительность (как метода так и прибора);
надёжность (как работы прибора так и проведенных измерений);
быстродействие.
На практике для выбора прибора с реализованным в нём методом применяется комплексный критерий качества, который может быть рассчитан по выражению 2.3.
К>кач> = к>1>*К>точн> + к>2>*К>надёжн> + к>3>*К>чувствит >+ к>4>*К>быстрод >, (2.3)
где: к>1>, к>2>, к>3>, к>4> – вес каждого критерия, их сумма ровняется единице;
К – базовый критерий, отражающий точность (надежность, чувствительность, быстродействие). Выбор базовых критериев осуществляется путём экспертных оценок, либо решением задачи оптимизации.
Точность прибора зависит от внутренних и внешних факторов, влияющих на измерительный процесс, рис.2.9.
Внешние факторы:
-
 состояние
коммуникаций;
состояние
коммуникаций;
-электрические и магнитные поля;
-персонал.
-

x
Внутренниефакторы
y
Температура
Давление
 Концентрация
Концентрация
Фазовое деление
Рис. 2.9. Факторы, влияющие на качество работы аналитического прибора
Температура является одним из главных управляющих воздействий на состояние объекта контроля. Она влияет на измерения характеристик состава и свойств веществ и выражается температурной погрешностью. Повышение точности измерения достигается за счёт компенсации температурной погрешности.
Учёт температурной погрешности в автоматизированных системах возможен аппаратными и программными средствами, которые разрабатываются после изучения влияния температуры на процесс измерения. С этой целью снимаются зависимости изменения косвенных параметров от температуры и строятся соответствующие графики.
На графике, представленном в виде прямой ( y = T ), влияние температуры на процесс измерения не отмечается, поэтому в структуре аналитического прибора система компенсации температурной погрешности не предусматривается.
Из анализа других графиков следует, что влияние температурной погрешности значительное, а значит, в цепь анналитического прибора должен встраиваться соответствующий компенсатор, учитывающий температурные условия протекания технологических процессов. Встроенный компенсатор перед началом измерений всегда настраивается первым до задания режима работы аналитического канала.
Для компенсации температурной погрешности в аналитических приборах применяются три способа: классический, эталонный, программный.
Классический способ устранения температурной погрешности состоит в измерении температуры и параметра x>i>, расчёте по математической модели погрешности ∆х ср. и значения параметра х ,выражение 2.3.
x = x>i> - ∆х ср (2.3)
Для реализации этого способа аналитический прибор содержит усилитель с переменным температурным резистором (рис. 2.11), который преобразует величину выходного сигнала.
При реализации этого способа в микропроцессорном аналитическом приборе в него встраивается термодатчик, преобразующий температуру в код F, рис. 2.11,а. Информация в цифровой форме заводится в микропроцессор (МП), в котором по математическим моделям рассчитывается измеренное значение x>i> и погрешность ∆х. Искомый параметр х отображается на индикаторе аналитического прибора. При наличии обратной связи погрешность используется для аппаратной компенсации измеренного значения.
Второй способ (эталонный) термокомпенсации заключается в использовании двухканальной структуры, один канал в ней является «эталонным» (сравнительным), рис. 2.12. Измеренные параметры по двум каналам сопоставляются между собой для исключения погрешности.
В микропроцессорном аналитическом приборе во второй канал включается первичный измерительный преобразователь с веществом, параметры которого нормированы.
Способ программного типа состоит из сопоставления исследуемого параметра с расчетом его по математической модели. При этом в математической модели могут использоваться температурные зависимости любого из ранее перечисленных способов (классического или «эталонные»), который подходит для данного процесса.
Для реализации третьего способа требуется достаточно мощное программное обеспечение, но он отличается точностью и скоростью получения результата.
3. Оптические методы анализа
Оптические методы аналитического контроля относятся к группе спектрометрических методов (см. тему 1). Они основаны на использовании известных законов распространения света – поглощения, рассеяния, свечения, преломления. Явления и эффекты, возникающие при взаимодействии анализируемого вещества, и электромагнитного излучения регистрируются электронными оптическими приборами – спектрофотометрами, фотоколориметрами, нефелометрами, флуориметрами, рефрактометрами, поляриметрами.
С помощью оптических методов определяются в лабораториях и контролируются на технологических линиях концентрации растворов различных веществ.
3.1 Общие сведения о спектроскопии
В основе спектроскопии лежит явление испускания электромагнитного излучения атомами или молекулами определяемого вещества.
Спектр электромагнитного излучения в зависимости от длины волн делят на ультрофиолетовую-180-400 нм (1 нанометр=10-9м), видимую-400-700нм, ближнюю инфрокрасную-700-1100нм области.
Электромагнитное излучение - свет - имеет двойственную природу - волновую и корпускулярную (волна - частица) и для его описания используют два вида характеристик - волновые и квантовые.
К волновым характеристикам относятся частота колебаний, длина волны, волновое число, а к квантовой характеристике относится энергия квантов,
Частота колебаний - ν - показывает число колебаний электромагнитного излучения (света) в 1 секунду, измеряется в с-1.
Длина волны λ - это путь, который проходит волной за время полного периода колебаний.



λ
Длина волны измеряется в метрах и его долях: сантиметрах – см; миллиметрах-мм; микронах-μ; миллимикронах – mμ; наномикронах - нμ (1нм =10-9 м = 10-7 см = 10-6 мм). Например, зеленый свет представляет собой электромагнитные излучения с длиной волны λ == 500 - 550 нм или 5,0 · 10 -5 - 5,5 · 10 -6 см.
Частота колебаний и длина волны связаны между собой выражением 3.1;
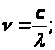 (3.1)
(3.1)
где: С - скорость света = 3 · 108 м/с = 3 · 1010 см/с
Величина, обратная длине волны называется волновым числом – ν и может быть рассчитана по выражению 3.2.
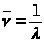 (3.2)
(3.2)
Для зеленого света волновое число составит
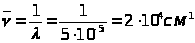
Если скорость света выражена в см/сек, длина волна в см - то частота колебаний будет выражена в герцах — Гц.
Для зеленого света:

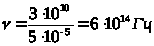
Энергия электромагнитного излучения определяется по выражению 3.3
E = h · ν , (3.3)
где h - постоянная Планка, равная 6,62 · 10-34 Дж с
3.2 Атомные спектры
Испускание света атомами происходит, за счет изменения энергии атомов. Атомы могут обладать только строго дискретными запасами внутренней энергии: Е>0>, Е>1>, Е>2> и т.д., т.е. атомы не могут иметь энергию, промежуточную между Е>0 >и Е>1> или между Е>1> и Е>2>. В невозбужденном, т. е. нормальном состоянии атомы обладают минимальной энергией Е>0>. При подведении энергии, достаточной для возбуждения атома — электроны атома переходят на более высокий энергетический уровень Е>1>, Е>2 >и т.д. и через очень короткое время ~ 10-8 с они самопроизвольно возвращаются в нормальное состояние и освобождающая при этом энергия излучается в виде светового кванта hν.
Совокупность излучаемых частот связана с энергетическими состояниями атома. Чем меньше длина волны, тем больше волновое число или частота, тем больше энергия электромагнитного излучения.
Наблюдаемые в природе электромагнитные излучения охватывают диапазон волн от десятков километров до тысячных долей ангстрема, распределение энергий излучения по длинам волн представляет спектр, который подразделяется на ряд областей, при взаимодействии с веществом излучение каждой области изменяет состояние молекулы по-разному. Это объясняется тем, что волны разных областей спектра имеют разную энергию, эта энергия действует на атом или молекулу, находящуюся в нормальном, невозбужденном состоянии и возбуждают их.
Характер спектров, наблюдаемых при взаимодействии электромагнитного излучения и строением энергетических уровней атомов и молекул исследуемых веществ, различен.
Основными характеристиками спектров является положение линий или полос, в шкале длин волн, а также их форма и интенсивность.
Положение спектральных линий и полос зависит от расстояния между энергетическими уровнями, переходы между этими уровнями обуславливают эти линии и полосы.
Строение энергетических уровней является индивидуальной характеристикой молекул (атомов, ионов) данного вещества, поэтому по положению тех или иных линий и полос в спектре можно судить о природе вещества, взаимодействующего с излучением.
Интенсивность спектральных линий и полос определяется тем, сколько квантов излучения данной частоты поглощается, испускается или рассеивается веществом в единицу времени, т.е. сколько молекул вещества участвуют в данном квантовом переходе. Это позволяет проводить количественные определения различных веществ по интенсивности линий и полос спектра.
Таким образом, действуя на вещество электромагнитным излучением, обладающим достаточной энергией, способной возбудить атомы - можно получить через короткий промежуток времени излучение в виде светового кванта hv (ΔЕ = hν )
Каждая спектральная линия отражает переход с одного энергетического уровня на другой.
Наиболее яркой в спектре будет линия, отвечающая переходу с первого возбужденного уровня на основной уровень. Линия, отвечающая этому переходу, называется резонансной. Например, у натрия
>11>Na Is22s22p63s1
|
↓ |
|||||
|
↑ ↓ |
↓ ↑ |
↓ ↑ |
↓ ↑ |
||
|
↑ ↓ |
При возбуждении атома натрия (нагревании, облучении и т.д.) валентный электрон (3s1) может переходить на уровни р и d, находиться на них очень короткое время и возвращаться вновь на основной. Этим переходам отвечают линии с длиной волны 588, 996 и 589, 593 нм. Это излучение окрашивает пламя в желтый цвет при введении солей натрия в пламя.
Это свойство атомов и ионов излучать свет в газообразном состоянии положено в основу методов эмиссионного и спектрального, где анализ основан на измерении длины волны, интенсивности и других характеристик излучений, испускаемых атомами за счет изменения их энергии.
Совокупность пространственно разделенных линий называют спектром.
Спектр, излучаемый раскаленными газами и парами, называется линейчатым или прерывистым, а спектр, который испускают раскаленные жидкие и твердые тела - сплошной.
Линейчатый спектр каждого элемента содержит ряд спектральных линий, соответствующих испускаемым лучам, характеризующихся определенной длиной волны λ или частотой колебания ν.
Наличие в спектре излучения таких линий дает возможность судить о наличии искомых элементов в исследуемом веществе, а интенсивность этих линий характеризует их количественное содержание. Цвет испускаемого или поглощаемого света зависит от длины волны. Например, наибольшая длина волны видимого света соответствует красному, а наименьшая – фиолетовому свету.
При проведении качественного спектрального анализа пользуются атласом спектральных линий.
В количественном анализе рассматривается связь между интенсивностью спектральной линии и концентрацией элемента в пробе.
3.3 Молекулярный спектр
Появление полос поглощения обусловлено дискретностью энергетических состояний частиц, которые поглощают энергию, а также от природы электромагнитного излучения. Интенсивно поглощаются кванты света, которые соответствуют энергии возбуждения частицы.
Любая молекула, в соответствии с квантовыми законами, является устойчивой в определенных стационарных состояниях. Переход молекулы из одного состояния в другое связан с получением и отдачей энергии (также как у атома).
Молекула сложная система, в молекуле имеют место различные виды движения составляющих ее частиц - колебательные и вращательные. Если молекуле сообщать разные количества энергии, действуя электромагнитным излучением, то каждому из этих количеств Е=hv - будут соответствовать различные виды спектров.
В отсутствии внешнего магнитного поля энергию молекулы можно представить выражением 3.4:
Е = Е>эл >+ Е>кол >+ Е>вр , (3.4)>
где:
Е>эл >— электронная энергия молекулы, обусловлена движением электронов, принимающих участие в образовании связей, так и локализованных вокруг ядра.
Е>кол> — колебательная энергия молекулы, обусловленная колебательным движением молекул, когда при неизменном положении центра тяжести молекул - периодически изменяется положение ядер и составляющих ее частиц.
Е>вр> — вращательная энергия молекулы, обусловленная вращательным движением молекулы, когда периодически происходит изменение ориентации молекулы в пространстве и ее частей относительно друг друга.
Электронная энергия значительно превышает колебательную, а колебательная – вращательную.
Е>эл>·>> Е>кол >> Е>вр>
По порядку величин отношение этих энергий составляет:
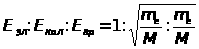 ,
,
где: m>е>- масса электрона;
М –масса молекулы .
Для большинства молекул m>е> =10-4 М = 10-5-10-5
Е>эл >: Е>кол >: Е>зр >= ~1 : 10-2 : 10-4
На основании этого можно представить энергетические уровни молекул, когда каждому электронному состоянию отвечает своя система колебательных уровней, а каждому колебательному своя система вращательных уровней:
 Е>эл>
Е>эл>
вр
кол
вр
кол

0
Е>эл>1
Чисто вращательные переходы, т.е. переход между вращательными уровнями соответствует наименьшему изменению энергии от единиц до сотен Дж /моль или 10-5 –10-3 Дж/моль.
ΔЕ>вр >= 10-5 · 10-3 Дж/моль
При этих переходах возникает чисто вращательный спектр, которому соответствует излучение микроволновой и части дальней ИК-области шкалы электромагнитных волн.
Переходам между колебательными уровнями одного и того же электронного состояния соответствует изменению энергии от единиц до сотен десятков КДж/м.
При этих переходах наблюдается колебательные спектры в ближней и дальней ИК - области.
Обычно при таких переходах изменяется и вращательная энергия молекул и происходит много переходов между вращательными подуровнями нижнего и верхнего колебательных уровней. В результате таких явлений в спектре возникает не одна линия, а совокупность близкорасположенных линий - образуя вращательную структуру колебательных полос (вращательно-колебательные спектры).
Переход молекулы из одного электронного состояния в другое составляет сотни КДж/моль, при этом возникают электронные спектры, наблюдаемые в видимой части спектра, а также в УФ - ближней и дальней.
Изменение электронного состояния молекулы сопровождается изменением колебательной и вращательной энергии, поэтому электронный молекулярный спектр состоит из совокупности колебательных полос, каждая из которых имеет вращательную структуру.
Из-за существования в молекуле переходов электронного, колебательного, вращательного - возникают и соответствующие спектры (молекулярные), они называются полосатыми.
Электронные спектры атомов газообразных веществ состоят из отдельных линий. Объясняется это тем, что атом не имеет колебательных и вращательных уровней энергии, а разрешенные значения электронной энергии - дискретны.
Спектры атомов более просты по сравнению со спектрами молекул.
Возвращение электрона в атоме из возбужденного состояния (с более высокого энергетического уровня на основной) в стабильное сопровождается выделением кванта энергии примерно равного поглощенному. Спектральные линии таких переходов лежат в области больших частот и малых длин волн.
Поглощение или испускание энергии можно определить по энергетическому состоянию молекулы в начальном и конечном энергетическом переходах, выражение 3.5.
ΔЕ = Е>1> - Е>2> = h · v, (3.5)
где: Е>1> — начальное состояние молекулы;
Е>2> — конечное состояние молекулы;
h — постоянная Планка (Дж/с);
v — частота излучения, поглощаемого или испускаемого при данном переходе (с-1). Если Е>2> > Е>1> - происходит поглощение излучения. Если Е>1>>Е>2> - происходит испускание (эмиссия)излучения.
Каждому переходу соответствует своя частота излучения и своя длина волны.
Каждое вещество обладает способностью поглощать лучистую энергию в виде квантов энергии, соответствующих определенным длинам волн.
В практической спектрофотометрии поглощение проводят' в ультрафиолетовой (200 - 400 нм), видимой (400 - 700 нм) и инфракрасной областях (700 - 2000 нм) спектра.
Спектрофотометрический анализ основан на определении спектра поглощения или измерении светопоглощения при строго определенной длине волны, которая соответствует максимуму кривой поглощения данного вещества.
Характер спектров, которые можно наблюдать при взаимодействии электромагнитного излучения с веществом, определяется энергией излучения и строением энергетических уровней молекул исследуемых веществ.
Основными характеристиками спектров является положение линий или полос в шкале длин волн, их форма и интенсивность.
3.4 Классификация оптических методов анализа
В оптических методах анализа используется зависимость между оптическими свойствами системы и её составом, рассматривается взаимодействие световой энергии (электромагнитного колебания) с веществом.
Поглощая электромагнитные излучения, атомы или молекулы переходят в новое состояние, возбуждённое, и избыточная энергия атомов и молекул может выделяться в виде вторичного излучения или расходоваться на повышение вращательной, колебательной и др. энергии.
В зависимости от вида частиц, поглощающих энергию и характера взаимодействия их с электромагнитным излучением, различают: атомно – абсорбционный анализ; молекулярно – абсорбционный анализ; флуориметрический (люминисцентный) анализ.
Атомно-абсорбционный анализ, основывается на том, что атом, поглощая подведённую энергию, переходит в возбуждённое состояние и примерно через 10-8 с спонтанно переходят в нормальное состояние электроны на нижележащие энергетические уровни, при этом происходит выделение (эмиссия) в виде дискретных и характеристических для каждого вида атомов электромагнитных колебаний в видимой, ультрафиолетовой или рентгеновской областях спектра. При этом спектры носят линейчатый характер. Характеристичность линейчатых спектров лежит в основе качественного эмиссионного спектрального анализа, а функциональная зависимость между концентрацией элемента в пробе и интенсивностью его спектральных линий положена в основу количественного анализа.
Молекулярно-абсорбционный анализ основан на поглощении электромагнитного излучения молекулами и сложными ионами анализируемого вещества в оптическом диапазоне спектра (ультрафиолетовой, видимой и инфракрасной областях ). В молекулярно-адсорбционной спектроскопии наблюдают и исследуют аналитические сигналы, вызванные электронными переходами внешних валентных электронов. Поглощение излучения в инфро - красной области, связанно с изменением вращения и колебания молекул. Это свойство молекул часто использует в целях идентификации различных соединений.
Анализ по поглощению и рассеиванию электромагнитного излучения взвешенными частицами анализируемого вещества подразделяется на турбидиметрию и нефелометрию. При прохождении света через дисперсную гетерогенную систему происходит ослабление светового потока в результате рассеивания и поглощения этого потока частицами дисперсной фазы, выражение 3.6.
J>0> = J>n> + J>р> + J , (3.6)
где:
J>0 >— интенсивность падающего светового потока;
J>n>> >— интенсивность поглощаемого светового потока;
J>р> — интенсивность рассеянного светового потока;
J — интенсивность прошедшего светового потока.
Турбидиметрия основана на измерении интенсивности светового потока, проходящего через дисперсную систему — J.
Нефелометрия основана на измерении интенсивности света, рассеянного дисперсной системой — J>р>.
Флуориметрический (люминесцентый) анализ, основан на измерении излучения, возникающего в результате выделения избытка энергии возбуждёнными молекулами анализируемого вещества.
Для возникновения явления люминесценции молекулы вещества облучаются и переводятся из основного в возбуждённое состояние. Энергия возбуждения должна быть достаточной для осуществления излучательного электронного перехода из возбуждённого состояния в основное. Это возможно для молекул с отрицательным устойчивым возбуждённым состоянием.
Фотометрия
Из методов молекулярного абсорбционного анализа наибольшее распространение получили фотометрические методы анализа — фотометрия. Они основаны на избирательном поглощении электромагнитного излучения молекулами анализируемого вещества.
В зависимости от используемой аппаратуры в фотометрическом анализе различают спектрофотометрический и фотоколориметрический методы анализа. Спектрофотометрический метод анализа заключается в поглощении монохроматического излучения, в котором все волны имеют одинаковую частоту — γ или длину волны — λ, а фотоколориметрический - поглощении полихроматического излучения.
Оба эти метода основаны на общем принципе существования пропорциональной зависимости между светопоглощением и концентрацией поглощающего вещества, являющегося однородной системой.
Любое вещество, способное отражать или поглощать электромагнитное излучение оптического диапазона (λ = 400 — 700 нм), имеет окраску. Непрерывное электромагнитное излучение в области длин волн 400 — 700 нм воспринимается глазом как белый цвет.
Окраска раствора обусловлена цветом той части светового потока (потока электромагнитного излучения), которая прошла через раствор непоглощённой. Визуально наблюдаемый цвет раствора является дополнительным к цвету поглощённого излучения.
Например, раствор, поглощающий жёлто-зелёную часть спектра, имеет длину волны λ = 560 — 570 нм, табл. 3.1.
Сущность фотометрии заключается в том, что определяемое вещество переводится в окрашенное состояние и с помощью оптического прибора определяется степень поглощения (электромагнитного излучения) окрашенным соединением, которая зависит от концентрации определяемого вещества. Основные оптические характеристики окрашенных растворов — цвет раствора и интенсивность окраски.
Фотометрический метод количественного анализа основан на способности определяемого вещества или его окрашенной аналитической формы поглощать электромагнитные излучения. Поглощение при определённой длине волны является материальным воплощением информации о качестве и количестве определяемого вещества, составляет аналитический сигнал. Возможность получения волны является материальным воплощением информации о качестве и количестве определяемого вещества, составляет аналитический сигнал. Возможность получения множества интенсивно окрашенных органических и неорганических соединений расширяют границы применения фотометрических определений в видимой области спектра с помощью довольно несложных и относительно недорогих приборов.
Таблица 3.1
Цвет раствора в зависимости от поглощённой части спектра
|
Спектральный диапазон поглощённой части, нм |
Цвет поглощённой части света |
Кажущийся цвет (дополнительный) |
|
400 - 450 |
Фиолетовый |
Жёлто-зелёный |
|
450 - 480 |
Синий |
Жёлтый |
|
480 - 490 |
Зелёно-синий |
Оранжевый |
|
490 - 500 |
Сине-зелёный |
Красный |
|
500 - 560 |
Зелёный |
Пурпурный |
|
560 - 575 |
Жёлто-зелёный |
Фиолетовый |
|
575 - 590 |
Жёлтый |
Синий |
|
590 - 625 |
Оранжевый |
Зелёно-синий |
|
625 - 750 |
Красный |
Сине-зелёный |
Фотометрические методы анализа высоко чувствительны и избирательны, а используемая в них аппаратура разнообразна. Эти методы широко применяются:
в системах автоматического контроля технологических процессов и готовой продукции;
при анализе исходных материалов в химической и металлургической промышленности, а также горных пород и природных вод;
при контроле продукции в сертификационных лабораториях,;
при экологической проверке состояния окружающей среды (воздуха, почвы, воды);
при диагностировании состояния людей и животных;
при определении примесей (10-4 – 10-6 %) в веществах высокой чистоты.
3.5.1 Основной закон светопоглощения — закон Бугера – Ламберта – Бера
Атом, ион или молекула вещества, поглощая квант света, переходит в более высокое энергетическое состояние. Обычно это — переход с основного, невозбуждённого уровня на один из более высоких уровней, чаще всего на первый возбуждённый уровень.
Если часть излучения поглощается веществом, то интенсивность излучения, по мере прохождения через слой вещества, падает.
Закон Бугера – Ламберта – Бера — основной закон светопоглощения связывает уменьшение интенсивности света, прошедшего через слой светопоглощающего вещества с толщиной его слоя и концентрацией в растворе.
Механизм поглощения монохроматического излучения, проходящего через стеклянный сосуд с раствором, проиллюстрирован на рис. 3.1.

J
 >0>
J>n>
J
>0>
J>n>
J
Рис. 3.1. Прохождение света через раствор, заключённый в стеклянный сосуд
При прохождении светового потока J>0> через слой раствора, заключённого в сосуд, его мощность ослабляется. К факторам, влияющим на ослабление светового потока, относятся:
отражение стенками сосуда - J>отр >;
поглощение окрашенным раствором - J>п>;
рассеивание взвесями, содержащимися в растворе - J>р.> Мощность выходящего из сосуда пучка света всегда будет меньше на величину потерь ( J>отр> + J>п> + J>р> ), выражение 3.7.
J = J>0> – ( J>отр> + J>п> + J>р> ) (3.7)
Ослабление светового потока происходит главным образом за счёт поглощения световой энергии раствором. В лабораторной практике при изучении поглощения света растворами пользуются одинаковыми кюветами, для которых мощность отражённой части светового потока заведомо известна, как правило, постоянна и настолько мала, что ею пренебрегают. При работе с истинными растворами достаточно чистых веществ потери мощности света за счёт рассеяния также незначительны, поэтому выражение 3.7 может быть записано более упрощённо (выражение 3.8).
J = J>0> - J>п >3.8
Мощность падающего светового потока J>0 >и прошедшего через раствор светового потока J могут быть измерены экспериментальным путём. Величина потерь рассчитывается по выражению 3.9.
J / J>0> = Т (3.9)
Отношение J / J>0> указывает на степень пропускания раствором светового потока и называется прозрачностью, а иногда пропусканием раствора. Коэффициент Т показывает, какая доля светового потока прошла через раствор, и принимает значение от 0 до 1.
Чем больше поглощается световой поток, тем меньше J по сравнению с J>0>, тем больше величина коэффициента Т.
Величина обратная прозрачности (выражение 3.10) называется непрозрачностью или поглощением раствора. Отношение мощности света, поглощенного раствором, к мощности падающего света ( J>n> / J>0> ), называется поглощающей способностью.
1 / Т = J>0> / J (3.10)
Логарифмированием выражения 3.10 рассчитывается оптическая плотность раствора (выражение 3.11). Она показывает степень поглощения излучения в зависимости от толщины слоя раствора и его окраски.
ℓg J>0> / J = Д = ℓg пL = L ℓg n , (3.11)
где: L – толщина поглощающего слоя;
ℓg n – постоянная величина, характерная для конкретного окрашенного раствора при прохождении через него света определённой длины;
Д – оптическая плотность (эту величину также называют абсорбционностью).
Выражение 3.11 отражает закон Бугера – Ламберта: слои вещества одинаковой толщины при прочих равных условиях всегда поглощают одинаковую долю падающего на них светового потока. Оптическая плотность вещества прямо пропорциональна толщине поглощающего слоя.
Позднее Бером было установлено, что поглощение света газами и растворами зависит от числа частиц в единице объёма, встречающихся на пути светового потока, т. е. от концентрации вещества в исследуемом растворе.
Закон Бугера – Ламберта – Бера устанавливает зависимость интенсивности поглощения света от концентрации вещества в растворе (С), толщины светопоглощающего слоя раствора(L) и молярного коэффициента поглощения света ( ε). Математическое выражение оптической плотности может быть представлено выражением 3.12. Оно получено экспериментальным путём, правильность его подтверждается с помощью математического аппарата.
Д = ε L С (3.12)
Объединённый закон Бугера – Ламберта – Бера является основным законом поглощения света растворами, он трактуется следующим образом: оптическая плотность раствора зависит от концентрации и природы исследуемого вещества, а также толщины слоя раствора, через который проходит световой поток (поток электромагнитных колебаний).
Для наглядности зависимость оптической плотности от концентрации вещества в растворе принято выражать графически, рис. 3.2. Она представлена прямой линий, идущей из начала координат и соответствует уравнению
D = k C ,где k = ε L ,а ε = k / 2,3.
Молярный коэффициент светопоглощения представляет оптическую плотность одномолярного раствора при толщине слоя светопоглощающего раствора 1 см.
ε = Д / LС (3.13)
Если С = 1 моль/л, L = 1 см, то Д = ε
Величина молярного коэффициента поглощения ε:
зависит - от длины волны проходящего света, температуры раствора и природы растворённого вещества;
не зависит - от толщины поглощающего слоя и концентрации растворённого вещества.
Д



α
Д

 >3>
>3>
tgα = ε

 Д>2>
Д>2>
Д >1>
>1>

C>1> C>2> C>3> C
Рис. 3.2. Зависимость оптической плотности от концентрации вещества
3.5.2 Молярный коэффициент светопоглощения
Молярный коэффициент светопоглощения отражает индивидуальные свойства вещества (окрашенного) и является их характеристикой. Для разных веществ он имеет различную величину. У слабоокрашенных веществ (например, хромат калия) молярный коэффициент светопоглощения составляет 400 – 500, а у сильноокрашенных (например, дитизонат цинка) - 94 000.
Следует иметь в виду, что значение молярного коэффициента поглощения, как правило, не превышает значения 100 000 – 120 000 для наиболее интенсивно окрашенных соединений. Его значение определяется экспериментально спектрофотометрическими методами.
Молярный коэффициент светопоглощения является характеристикой чувствительности фотометрических реакций, чем больше его величина, тем чувствительнее и точнее определение. При выборе реактивов, дающих цветовую реакцию с определяемым веществом, выбирают тот, который образует соединения с максимальным коэффициентом светопоглощения.
Из закона Бугера–Ламберта–Бера вытекают два вывода, которые имеют практическое значение.
Первый вывод. При одинаковой интенсивности окраски одного и того же вещества их концентрации обратно пропорциональны толщине поглощающих слоёв.
Доказательство. Предположим, что имеются два раствора одного и того же вещества, но с разной концентрацией. Согласно закону Бугера-Ламберта-Бера (см. выражение 3.11) оптическая плотность (Д) каждого раствора может быть представлена следующими математическими выражениями:
ℓg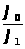 = εL>1>C>1>
ℓg
= εL>1>C>1>
ℓg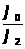 = εL>2>C>2>
= εL>2>C>2>
Принимая во внимание, что исследуемые растворы одинаково освещены, т. е. на них воздействует световой поток интенсивностью равной J>0. >Выравнивание световых потоков (J>1> = J>2>), прошедших через растворы может быть достигнуто подбором толщин просвечиваемых растворов L>1> и L>2.> Исходя из этого, имеют место следующие равенства:
ℓg
= ℓg
следовательно εL>1>C>1>
= εL>2>C>2>,
а так как ε>1>
= ε>2> тогда
L>1>C>1>
= L>2>C>2.>
Таким образом — при одинаковой интенсивности окраски одного и того же вещества их концентрации обратно пропорциональны толщине поглощающих слоёв.
Второй вывод. При условии равенства толщин исследуемого раствора и стандартного раствора одного и того же вещества (L>1> = L>2>) зависимость между их оптической плотностью и концентрацией прямопропорциональна:
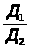 =
=
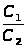
Оптическая плотность раствора, содержащего несколько окрашенных веществ, обладает свойством аддитивности, которое называют законом аддитивности светопоглощения (аддитивность-лат. additio прибавление-результат получаемый путём сложения). В соответствии с этим законом поглощение света, каким - либо веществом не зависит от присутствия в растворе других веществ, так как каждое из окрашенных веществ будет вносить свою величину в экспериментально определяемую оптическую плотность — Д.
Д = Д>1> + Д>2> + Д>3>, т. к. L-const, то имеет место сумма (ε>1>C>1> + ε>2>C>2> + ε>3>C>3>)
3.5.3 Спектры поглощения
Все окрашенные соединения характеризуются избирательным поглощением света.
Для характеристики окрашенных растворов различных окрашенных соединений пользуются их спектрами поглощения — кривыми светопоглощения, которые определяют зависимость оптической плотности Д или молярного коэффициента поглощения ε от длины волны λ или частоты γ
Д = f(λ) Д = f(γ)
ε = f(λ) ε = f(γ)
Для получения такого спектра (кривой светопоглощения) в таких координатах — проводят серию измерений оптической плотности или молярного коэффициента светопоглощения при различных длинах волн, измерение проводится вначале через 10 – 20 нм, а после границы максимума измеряют через 1 – 2 нм.
Поглощение света измеряют в оптическом диапазоне спектра в ультрафиолетовой (185 – 400 нм), видимой (400 – 760 нм) и инфракрасной (760 – 1000 нм) областях спектра. Кривые светопоглощения снимают с помощью спектрофотометров, рис 3.3.
У окрашенных веществ максимум поглощения света, в большинстве случаев, находится в видимой области спектра (≈ 500 нм), но не может быть смещен в ультрафиолетовую область (K>2>CrO>4>), а также может смещаться и в инфракрасную — (CuSO>4>).
Спектры поглощения позволяют выбрать оптимальную длину волны для аналитических измерений. Максимуму спектра поглощения соответствует максимальное значение молярного коэффициента поглощения — Еmax, т.е. максимальной чувствительности.
 Д
3
Д
3

 1,4
max
1,4
max
—
—
 1,0
— 1
1,0
— 1
 —
—
—
—
 — 2
— 2


 •
• • • • •
•
• • • • •
0 100 200 300 400 500 λ
Рис. 3.3. Спектры поглощения водных растворов хромата (1), дихромата (2) и перманганата (3) калия
Величина Д = ℓg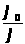 характеризует поглощательную способность
вещества, называемую поглощением или
светопоглощением — эту величину снимают
со шкалы прибора при аналитических
определениях. Иногда шкала колибруется
на пропускание — Т, %.
характеризует поглощательную способность
вещества, называемую поглощением или
светопоглощением — эту величину снимают
со шкалы прибора при аналитических
определениях. Иногда шкала колибруется
на пропускание — Т, %.
Между оптической плотностью Д и пропусканием Т существует связь, выражение 3.14.
Т =
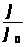 · 100
=
· 100
=
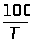
ℓg
= ℓg100
- ℓgТ
ℓg
= Д Д = ℓg100
- ℓgТ
= 2 - ℓgT
Д = 2 - ℓgT (3.14)
Зависимость оптической плотности от концентрации выражается графиком, рис.3.2.
Тангенс угла наклона (α) градуировочного графика к оси (С) указывает на чувствительность метода. Чем больше угол наклона к оси концентрации градуировочного графика, тем более чувствителен метод определения.
На основании закона Бугера – Ламберта – Бера можно определить нижнюю границу диапазона содержания определяемых веществ (Сmin)
Д>min>
= E>λ>
· L
· C>min>,
если L
= 1 см С>min>
=
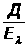
Использование закона Бугера – Ламберта – Бера позволяет проводить различные расчёты на основании фотометрических измерений и определений.
Пример: Вычислить молярный коэффициент поглощения железа в растворе, содержащем 0,0028 г Fe в 500 мл раствора, при L = 4 см, если Д = 0,28.
Приводит концентрацию к системе моль/л.
 0,0028
г — 500 мл
0,0028
г — 500 мл
Х — 1000 Х = 0,0056 г/л
Fe / 56
56
г — 1 моль
0,0056 — Х Х =
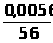 = 10-4 моль/л
= 10-4 моль/л
Д = ε · L
· C
ε
=
 = 700
= 700
Пример: Вычислить концентрацию ионов железа [Fe3+] в мг/л в промышленной воде, если после обработки 100 мл этой воды получено 25 мл окрашенного раствора с оптической плотностью Д = 0,46 при L = 1 см и ε = 1100.
1. Д = ε
· L
· C
C= =4,18·10-5
моль/л
=4,18·10-5
моль/л
4,18·10 –5 - 1000 мл
Х
25 мл Х= =
0,104·10-5моль
=
0,104·10-5моль
56 г - 1 моль
Х
- 0,104·10 –5
Х=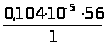 =5,85·10-5
г в 25 мл
=5,85·10-5
г в 25 мл
5,85 · 10-5 ————— 100 мл
Х ————— 1000 мл Х = 5,85 · 10-4 г/л = 5,85 · 10-1 мг/л = 0,585 мг/л
По данным фотометрических определений можно найти молярный коэффициент светопоглощения, концентрацию ( %, моль/л, титр) и др. величины.
При работе с разбавленными окрашенными растворами измерение их оптической плотности следует производить в той области спектра, где поглощение лучей максимально. Это позволит провести количественное определение с наибольшей точностью и чувствительностью.
Рассмотрим точность измерений оптической плотности окрашенного раствора на разных участках видимой области спектра.
Обычно вещества максимально поглощают лучи λ ε= 550 нм и минимально при λ = 640 нм.
Рассмотрим, как изменяются оптические плотности трёх растворов с разными концентрациями С>1>, С>2>, С>3>, причём С>1>>C>2>>C>3>, при λ>max> и λ>min>, построим график, рис. 3.4.
При изменении концентрации вещества в интервале ΔС изменение оптической плотности ΔД при λmax будет значительно больше, чем при λmin, это обуславливает наименьшую погрешность измерения, т.е. наибольшую точность.
Спектр поглощения характеризует зависимость оптической плотности (или молярного коэффициента поглощения) от длины волны.
Область максимального поглощения лучей характеризуется также размытостью максимума поглощения — интервалом длин волн (λ>1/2>>max> — λ>1/2>>min>) отвечающим половинным значениям максимального молярного коэффициента поглощения или максимальной оптической плотности раствора. Максимум поглощения света в определённой области является важной оптической характеристикой.
 Д
Д
max
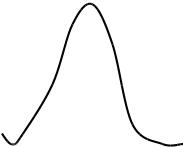




λ, нм
Д Д
Д


 C>1>
tgα
C>1>
tgα
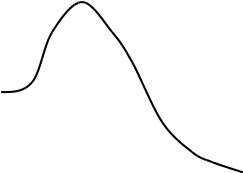
 ●
●
●
●

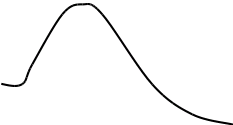
 ●C>2>
●
●C>2>
●
 C>3>
ΔД
C>3>
ΔД
 ●
● tgα
●
● tgα


 ●
●
●
●
 ●
ΔД ●
●
ΔД ●

 ●
● ●
●
● ●
 550
650 λ
С>3> С>2>
ΔС
С>1> С
550
650 λ
С>3> С>2>
ΔС
С>1> С
λmax = 550 λmin = 650 при λmax — ΔД> в интервале ΔС
Рис. 3.4. Графики, отображающие зависимость оптической плотности растворов от их концентрации
Спектр поглощения характеризуется наличием в нём определённого числа полос. Каждая полоса характеризуется положением максимума и выражается соответствующей длиной волны λmax, высотой — Дmax, или Еmax и полушириной, т.е. расстояние между длинами волн, соответствующим половинным значениям максимальной оптической плотности λ>1/2>>max> — λ’ >1/2>>max>
Кривые спектров поглощения позволяют выбрать оптимальную длину волны при аналитических исследованиях.
3.5.4 Взаимодействие света с дисперсными гетерогенными системами
Некоторые элементы не дают окрашенных аналитических форм, или образуемые соединения не достаточно устойчивы. Поэтому фотометрическое определение таких компонентов не проводится, а используется способность таких веществ образовывать достаточно устойчивую дисперсную систему (взвесь мельчайших твёрдых частиц в растворе). Например, это относится к определению Cl-, SO>4>2-, C>2>O>4>2- и др. ионов, которые образуют осадки. Для предотвращения коагуляции частиц в дисперсной системе (суспензии) вводятся стабилизирующие коллоиды (желатин, крахмал и др.).
Ag+ + Cl- → ↓AgCl Образовались белые
Ba2+ + SO>4>2- → ↓BaSO>4> осадки гетерогенных
Ca2+ + C>2>O>4>2- → ↓CaC>2>O>4> систем.
При прохождении света через дисперсную гетерогенную систему происходит ослабление светового потока в результате рассеивания и поглощения его частицами дисперсной фазы. Интенсивность рассеяния возрастает с увеличением числа рассеивающих частиц
J>0> = J>n> + J>p> + J
Это явление используется в турбидиметрических и нефелометрических методах для качественной и количественной оценки анализируемых веществ, рис. 3.5
J>n>


 J
J

 J>0>
J>0>
J>р>
Рис 3.5. Схема .измерения световых потоков в турбодиметрии и нефелометрии
Турбидиметрия основана на измерении интенсивности светового потока, прошедшего через дисперсную систему — J.
Нефелометрия основана на измерении интенсивности светового потока, рассеянного дисперсной системой — Jр.
Турбидиметрия и нефелометрия подчиняются некоторым закономерностям, которые перестают действовать, когда размеры частиц дисперсной системы приближаются к длине волны падающего света.
В турбидиметрии пользуются соотношением, аналогичным закону Бугера –Ламберта – Бера, с заменой коэффициента светопоглощения на коэффициент мутности, выражение 3.15.
Д = ℓg
= t
L ,
(3.15)
где: t — коэффициент мутности;
L — толщина слоя.
Коэффициент мутности, это величина, обратная толщине такого поглощающего слоя, которая уменьшает интенсивность падающего светового потока в 10 раз.
В нефелометрии измеряют интенсивность светового потока, который дисперсная система рассеивает (Jр), а способность частиц к рассеиванию определяется размером частиц и длиной волны падающего света, что выражается уравнением Рэлея (выражение 3.16).
J>p>
= J>0
>[F( ) · (1 + соsQ)]
, (3.16)
) · (1 + соsQ)]
, (3.16)
где: F
- функция от показателей преломления F
= n>1>2
-
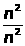 ;
n —
коэффициент преломления растворителя;
;
n —
коэффициент преломления растворителя;
n>1> — коэффициент преломления частиц;
N — общее число частиц в данном растворе;
V — объём частиц взвесей, рассеивающих свет;
λ — длина волны падающего света;
R — расстояние от детектора (до наблюдателя);
Q — угол рассеивания между падающим и рассеянным потоками.
Если определяется только размер частиц и их концентрация, то измеряется интенсивность рассеянного света под одним углом. В этом случае уравнение Рэлея представляется в виде:
J>p> = J>0> · k · c · V
Градуировочный график в нефелометрии строят в координатах Jр — С.
Мутность дисперсной системы, в соответствии с уравнением Рэлея, можно выразить коэффициентом мутности или коэффициентом светопоглощения :
t
=
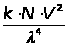 , Д =
, Д =
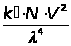
Если взять отношение оптических плотностей для двух дисперсных систем малорастворимых веществ с одинаковым размером частиц, оно будет равно отношению концентраций, а при одной и той же концентрации отношение оптических плотностей пропорционально размерам частиц.
Используя метод нефелометрии можно определить содержание сульфат-ионов, содержание хлорид-ионов и др. ионов в приготовленных растворах, а также в природных веществах.
Турбодиметрические и нефелометрические определения обладают чувствительностью соизмеримой с фотометрическими определениями. Эти методы в практике производственных лабораторий применяют ограниченно, т. к. трудно получить одинаковые по размерам частицы взвеси. Их, как правило, заменяют фотометрическими и электрометрическими методами.
3.5.5 Роль химической реакции, используемой в фотометрическом анализе
Химические реакции, используемые в фотометрическом анализе, несмотря на различие в их химизме, должны обязательно сопровождаться возникновением, изменением или ослаблением светопоглощения (цвета) раствора. Каждая цветная реакция должна протекать избирательно, быстро, полностью, строго по уравнению и в соответствии с законами стехиометрии.
Кроме того, окраска образующейся аналитической формы должна быть устойчивой во времени к действию света и других внутренних и внешних факторов. В тоже время, светопоглощение раствора, несущее информацию о концентрации поглощающего вещества, должно подчиняться законам, связывающим светопоглощение и концентрацию вещества в поглощающем растворе.
В неорганическом фотометрическом анализе наиболее часто используют реакции комплексообразования ионов определяемых элементов с неорганическими и органическими реагентами, реже реакции окисления-восстановления, синтеза и других типов.
В органическом фотометрическом анализе чаще применяют реакции синтеза окрашенных соединений, которыми могут быть азосоединения, полиметиловые и хинониминовые красители, отдельные представители нитросоединений и др. Иногда используют собственную окраску вещества.
При фотометрических определениях в результате аналитической реакции получают окрашенное соединение, которое можно считать удобным для применения, если оно имеет постоянный состав, отвечающий определённой химической формуле.
Постоянный состав окрашенного соединения обуславливает постоянство интенсивности окраски раствора и является одним из основных факторов, влияющих на точность фотометрического определения. Однако на практике этот принцип нарушается по нескольким причинам:
а) Непостоянство состава окрашенного комплекса в связи со ступенчатым характером его образования и диссоциации.
Например, ион Fe3+ образует с SCN- ряд комплексных ионов кроваво-красного цвета различной интенсивности в зависимости от избыточной концентрации [SCN-], моль/л.
[SCN-] = 5 · 10-3 Fe3+ + SCN- = [FeSCN]2+
[SCN-] = 1,2 · 102- Fe3+ + SCN- = [Fe(SCN)>2>]+
[SCN-] = 4 · 10-2 Fe3+ + SCN- = [Fe(SCN)>3>]0
[SCN-] = 1,6 · 10-1 Fe3+ + SCN- = [Fe(SCN)>4>]-
[SCN-] = 7 · 10-1 Fe3+ + SCN- = [Fe(SCN)>5>]2-
Чтобы избежать больших ошибок из-за непостоянства интенсивности окраски анализируемых растворов, необходимо выбирать такие реагенты, с которыми определяемый ион давал бы прочное комплексное соединение, состоящее из одного комплексного иона.
Если такой реагент выбрать невозможно, то определение следует проводить при избыточных, но одинаковых концентрациях реагента в стандартном и исследуемом растворах. Несоблюдение этого условия приводит к получению окрашенных растворов различной интенсивности и к ошибкам.
б) Разложение окрашенного соединения во времени.
Многие окрашенные соединения изменяют, интенсивность своей окраски во времени. Иногда, скорость реакции мала и образование окрашенных соединений происходит не сразу, а по истечении некоторого времени — (10-20 мин) достигает максимального и постоянного значения.
В других случаях образование окрашенного соединения происходит очень быстро, но спустя некоторое время интенсивность окраски начинает уменьшаться и может вообще обесцветиться. Это может произойти по причине окислительно-восстановительных реакций между реагирующими ионами, либо окрашенное соединение разрушается под влиянием присутствующих в растворе посторонних веществ, изменение рН среды, явлений ассоциации, ротолиза и др.
В фотометрическом анализе можно использовать только такие окрашенные соединения, которые сохраняют устойчивую окраску не менее 10-15 минут.
Иногда к исследуемому окрашенному раствору добавляют стабилизаторы — желатин, крахмал, гуммиарабик и др.
Если нет сведений об изменении интенсивности окраски во времени каких-то соединений, применяемых в фотометрическом анализе — можно получить такие сведения практически. Для этого нужно приготовить 2-3 пробы окрашенного соединения и проследить за изменением интенсивности его окраски в течение времени сравнивая со свежеприготовленными растворами той же концентрации визуально, или измерив оптическую плотность.
в) Изменение состава окрашенного комплекса по причине присутствия посторонних веществ, взаимодействующих с определяемым ионом или выбранным реагентом.
Посторонние ионы, присутствующие в анализируемом растворе одновременно с определяемым ионом часто оказывают значительное влияние на результаты фотометрического анализа.
Например, при определении Fe3+ присутствие небольших количеств фторид-ионов вызывает заметное обесцвечивание раствора роданида железа (Кр = 5,2 · 102), так как ионы железа связываются в более прочный фторидный комплекс (Кр = 1,6 · 10-5) и не при каких значениях рН раствора влияние фторид-ионов устранить не удаётся.
В присутствии фторид-ионов Fe3+ следует определять с помощью другого реагента, например, салициловой кислоты. Она при взаимодействии с Fe3+ образует более прочный салицилатный комплекс, что устраняет мешающее действие фторид-ионов.
Влияние рН на окрашенные комплексы выражается в различных формах, но чаще всего сводится к разрушению или изменению состава окрашенного соединения.
Иногда оно способствует образованию окрашенных комплексов с посторонними ионами, присутствующими в растворе, а также обуславливает изменение растворимости окрашенных соединений и влияет на состояние окислительно-восстановительного взаимодействия.
3.5.6 Классификация приборов для фотометрических измерений
Приборы в фотометрических измерениях и определениях предназначены для разложения электромагнитного излучения оптического диапазона на монохроматические составляющие с последующим измерением оптической плотности растворов. К ним относятся фотометрические приборы - фотоэлектроколориметры и спектрофотометры. В этих приборах аналитическим сигналом является светопоглощение анализируемого раствора.
Фотометрические приборы, применяемые для измерения величины светопоглощения растворов, классифицируются по следующим признакам, рис. 3.6:
1.По способу регистрации измерений — регистрирующие и нерегистрирующие.
2.По способу разложения излучения, т.е. по способу монохроматизации лучистого потока (призменные, дифракционные). Приборы, в которых монохроматизация достигается с помощью светофильтров, называют фотоэлектрополяриметрами. Приборы, позволяющие достигать высокую степень монохроматизации светового потока, называют спектрофотометрами.
3. По назначению — для эликсионного анализа, для абсорбционного анализа.
4 .По области, в которой работает прибор — инфракрасной, видимой, ультрафиолетовой.
5. По устройству. Однолучевые приборы - с прямой схемой измерения. Двулучевые приборы - с компенсационной схемой измерения.
3.5.7 Фотоэффект и фотоэлементы
При измерении оптической плотности сравнивают и оценивают различие потоков света: (J>0>) – направленного на кювету с анализируемым раствором и (J) – прошедшего через раствор.
На практике величину светопоглощения анализируемого раствора измеряют относительно раствора сравнения, который является эталонным. Светопоглощение эталонного раствора, принимается за оптический нуль. Интенсивность регистрируемые потока, проходящего через анализируемый раствор и раствор сравнения, измеряют фотоэлектрическим способом, после преобразования светового электромагнитного излучения в электрический сигнал.
В качестве устройства для измерения плотности светового потока, прошедшего через раствор, используется фотоэлемент. В фотоэлементе энергия электромагнитного излучения преобразуется в электрическую энергию, которая в последующем регистрируется электроизмерительным прибором.
Преобразование энергии электромагнитного излучения в электрическую энергию в фотоэлементе происходит из-за отрыва электронов от атомов различных веществ под воздействием световой энергии. Это явление называется фотоэффектом.
Согласно закону Столетова фототок прямо пропорционален световому потоку, выражение 3.17. Чем больше световой поток, тем больше квантов энергии электромагнитного излучения попадает на поверхность металла, тем большее число электронов освобождается и тем больше будет фототок. Чем больший фототок даёт фотоэлемент, тем он чувствительнее.
I = k J, (3.17)
где: I – фототок, мкА;
k - коэффициент пропорциональности;
J - мощность светового потока, лк (люкс-единица освещённости, в СИ обозначается lx).
Различают два вида чувствительности: общую (интегральную) и спектральную (цветовую). Общая чувствительность фотоэлементов определяется по отношению к свету, излучаемому обыкновенными электрическими лампами накаливания с вольфрамовой нитью. Спектральная чувствительность фотоэлементов – это их чувствительность к свету различных длин волн.
Для измерения мощности световых потоков применяют два типа фотоэлементов:
1) основанных на внешнем фотоэффекте (вакуумные фотоэлементы);
2) основанных на фотоэффекте в запирающем слое («вентильные» фотоэлементы).
Фотоэлементы, основанные на внешнем фотоэффекте, представляют собой вакуумный стеклянный сосуд. На внутреннюю поверхность одной из стенок сосуда наносится щелочной металл, который выполняет функцию фотокатода. В отдельных случаях функцию фотокатода выполняет размещенная внутри металлическая пластинка.
Внутри сосуда перед катодом располагается анод. Он предназначен для сбора электронов, выбиваемых световым потоком из катода.
В фотоэлектроколориметрах и спектрофотометрах используют, как правило, сурьмяно-цезиевые фотоэлементы. Сурьмяно-цезиевые фотоэлементы отличаются высокой разрешающей способностью. Они высокочувствительны во всех областях спектра, очень хорошо работают до температуры равной 500С. Однако при повышении температуры вносятся большие погрешности, для их устранения в современных приборах применяются специальные устройства.
В фотоэлементах с запирающим слоем использована способность полупроводников к внутреннему фотоэффекту, т. е. возникновению тока под действием света на границе между полупроводником и металлом. Чувствительность этих фотоэлементов невелика и равна 540 – 560 нм ( как у человеческого глаза) и зависит от способа обработки поверхности фотоэлемента. Нечувствительность к ультрафиолетовому излучению ограничивает применение селеновых фотоэлементов.
3.5.8 Общие принципы устройства фотометрических приборов
Фотометрические приборы в зависимости от числа используемых при измерении фотоэлементов делятся на две группы:
приборы с одним фотоэлементом (однолучевые или одноплечие приборы);
приборы с двумя фотоэлементами (двулучевые или двуплечие приборы).
А. Однолучевой фотометрический прибор
В нём все основные узлы расположены на одной линии и световой поток (электромагнитное излучение) идёт одни пучком от источника света.
Принципиальная схема однолучевого прибора с прямым способом измерения (КФК-2, КФК-3, СФ-46) представлена на рис. 3.8.
Перед проведением фотометрического измерения в приборе устанавливают необходимый светофильтр (определённую длину волны). Проверяют настройку прибора на электрический нуль.
В световой поток устанавливают кювету с раствором сравнения 4.
Электронным усилителем 6 усиливают фототок с помощью вспомогательной диафрагмы, устанавливают стрелку указывающего прибора на отметку 100 %-ного пропускания, что соответствует оптическому нулю.
Вместо кюветы с раствором сравнения в световой поток помещают кювету с анализируемым раствором, при этом световой поток уменьшается пропорционально его концентрации (в соответствии с законом Бугера – Ламберта – Бера). Стрелка показывающего прибора зарегистрирует величину, соответствующую пропусканию анализируемого раствора. На шкале такого прибора имеется шкала, показывающая степень пропускания, а также логарифмическая шкала оптических плотностей. При необходимости показание по шкале пропускания можно пересчитать на поглощение, используя следующие выражения:
Т =
· 100 Д =
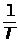 · 100 = 2 - ℓgTНа
· 100 = 2 - ℓgTНа
Оптическую плотность (пропускание) измеряют относительно эталона, пропускание которого принимают за 100%, а оптическую плотность — равной нулю.
Б. Двулучевой фотометрический прибор
В двулучевом фотометрическом приборе световой поток делится на два, которые идут параллельно друг другу, рис.3.9. Один поток идёт через кювету с анализируемым раствором, а второй через кювету со стандартным (нулевым) раствором.
На этой основе работают оптические приборы КФК-2 и КФК-3 (колоримерт фотоэлектрический концентрационный). Эти приборы предназначены для измерения в отдельных участках диапазона длин волн 315 – 980 нм, выделяемых светофильтрами, коэффициентов пропускания и оптической плотности растворов и твёрдых тел, а также определения концентрации веществ в растворах методом градуировочного графика.
В схеме КФК-2 на пути светового потока имеется пластина, которая разделяет поток на две части, 10% потока идёт на фотодиод (ФД-7к) при измерениях в области спектра 590 – 980 нм и 90% — на фотоэлемент (Ф-2в) при измерении в области спектра 315 – 540 нм.
Регистрирующим устройством служит микроамперметр типа М-907, имеющий шкалу деления от 0 до 100, соответствующую шкале пропускания Т. (Её следует пересчитать на поглощение). Если показания регистрирует микроамперметр М-907-10, то он даёт показание в делениях пропускания и оптической плотности определения концентрации веществ в растворах методом градуировочного графика.
В схеме КФК-2 на пути светового потока имеется пластина, которая разделяет поток на две части, 10% потока идёт на фотодиод (ФД-7к) при измерениях в области спектра 590 – 980 нм и 90% — на фотоэлемент (Ф-2в) при измерении в области спектра 315 – 540 нм.
Регистрирующим устройством служит микроамперметр типа М-907, имеющий шкалу деления от 0 до 100, соответствующую шкале пропускания Т (Её следует пересчитать на поглощение). Если показания регистрирует микроамперметр М-907-10, то он даёт показание в делениях пропускания и оптической плотности определения концентрации веществ в растворах методом градуировочного графика.
Спектрофотометр СФ-46 представляет собой однолучевой прибор со встроенной микропроцессорной системой. Он предназначен для измерения оптической плотности и пропускания жидких и твёрдых веществ в диапазоне волн 190 – 1100 нм.
Диспергирующим элементом служит дифракционная решётка. Световой поток после прохождения всей цепи устройств собирается в один из фотоэлементов — сурьмяно-цезиевый (186 – 650 нм) или кислородно-цезиевый (600 – 1100 нм). Источниками излучения служит дейтеривая лампа (186 – 350 нм) и лампа накаливания (320 – 1100 нм).
В данном случае фотоэлементы подключены по дифференциальной схеме (токи от фотоэлементов идут навстречу друг другу). На такой основе работают ФЭК-56, ФЭК-57 (фотоэлектрический колориметр-нефелометр), световой поток от источника света после светофильтра делится на два равных потока — левый и правый, а далее через систему зеркал и линз попадают на кюветы с растворами (анализируемым и стандартным). На пути правого потока щелевая диафрагма (измерительная), она связана со шкалой барабана. На пути левого потока также имеется щелевая диафрагма, служащая для ослабления светового потока, падающего на левый фотоэлемент.
На пути светового потока с помощью специальной рукоятки устанавливаются светофильтры, табл. 3.1.
На измерительных барабанах нанесены две шкалы: чёрная — процент пропускания, красная — оптическая плотность. Измерение начинают спустя 20 мин. после включения прибора.
Таблица 3.1
Эффективная длина волны светофильтров
|
Номер светофильтра |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Эффективная длина волны |
315 |
364 |
400 |
440 |
490 |
540 |
582 |
610 |
639 |
Для проведения измерений при перекрытых световых потоках на пути левого потока устанавливают кювету со стандартом, а на пути правого — кювету с исследуемым раствором и правый барабан устанавливают на полное пропускание — 100%.
Вращением левого измерительного барабана добиваются смыкания сектора индикаторной лампы. Затем на пути правого потока устанавливают кювету со стандартом и вращением правого барабана снова добиваются смыкания сектора индикаторной лампы. Оптическую плотность отсчитывают по шкале правого барабана.
В. Основные узлы приборов, к ним относятся:
Источники излучения — чаще ртутно-кварцевая лампа, галогеновая, водородная, дейтеривая;
Светофильтры;
Кюветы;
Фотоэлементы.
Кроме этого, в зависимости от конструкции и типа прибора, могут входить поворотные зеркала, призмы, дифракционные решётки, диафрагмы и т.д.
Светофильтры — это специальные устройства, выполненные из окрашенного прозрачного материала, чаще цветного стекла, которое используется для регулировки длин волн, получения монохроматического излучения. Для каждого анализа светофильтр выбирают опытным путём, для этого измеряют оптическую плотность с различными светофильтрами одного и того же раствора и строят кривую зависимости оптической плотности (Д) от длины волны (λ).
Д
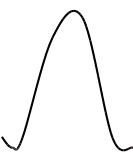
 max
max

λmax λ
Выбирают светофильтр такой длины волны, при котором поглощение света раствором max, когда светофильтр пропускает максимальное количество света.
Кюветы — сосуды, изготовленные из прозрачного материала, в которые помещают исследуемый раствор. На рабочей поверхности кюветы указывается толщина слоя с точностью до 0,001. Выбор кюветы осуществляется опытным путём, для этого измеряют оптическую плотность одного и того же раствора в кюветах разной толщины и выбирают ту, для которой оптическая плотность (Д) приближена к 0,4. Д = 0,4, т.к. шкала прибора на разных участках имеет разную относительную ошибку, а в области Д = 0,4 эта ошибка минимальна.
Например, экспериментально найдены результаты:
|
Толщина к4юветы, мм |
5 |
10 |
20 |
30 |
50 |
|
Оптическая плотность |
1,12 |
0,38 |
0,65 |
1,02 |
1,30 |
Следовательно, для данного определения целесообразнее выбрать: h = 10 мм
Конденсоры — устройства, которые представляют линзу или систему линз, позволяющие направлять световой поток параллельным пучком


Фотоэлементы, устройства, предназначенные для перевода световой энергии в электрическую, см. раздел 3.5.7.
3.6 Рефрактометрический метод анализа
Метод, основанный на измерении показателя преломления светового потока при прохождении его через анализируемый раствор, называется рефрактометрическим. Он широко применяется как в лабораторной, так и в промышленной практике.
С помощью рефрактометрического метода быстро определяют концентрации водных, спиртовых эфирных и других растворов. Им пользуются в лабораториях и автоматизированных линиях аналитического контроля химической нефтехимической, фармацевтической и пищевой промышленности. Его применяют при идентификации и установлении чистоты толуола, бензола, керосина, водно-спиртовых смесей, сахара, вина, а также при аналитическом контроле производства синтетического каучука, волокон, пластмасс и др. продукции.
3.6.1 Теоретические основы метода
При переходе луча света из одной прозрачной среды в другую, направление его меняется, рис. 3.10. Это явление называется преломлением.
Известно, что при прохождении света через оптически более плотную среду его скорость уменьшается. Замечено, что при этом угол падения луча при выходе из среды изменяется. При переходе луча из среды менее оптически плотной в среду более оптически плотную угол падения луча (α) больше угла преломления (β), таким же образом изменяется и скорость распространения световых волн.
Отношение синуса угла падения к синусу угла преломления называется относительным показателем преломления второй (анализируемой) среды относительно первой (эталонной), выражение 3.6.1.
Sin α / sin β = v>1> / v>2> = n (3.6.1)
Показатель преломления зависит от природы вещества, температуры и длины волны света.. Например, для температуры 200 С и длины волны 589 нм показатели преломления п некоторых веществ имеют следующие значения: стекло 1,5 – 1,9; алмаз – 2,42; плавленый кварц – 1,46; кристаллический кварц – 1,54; глицерин – 1,47; этиловый спирт – 1,36; вода – 1,3330 (при 150С – 1,3395, при 250С - 1,3325). Поэтому при точных измерениях показателя преломления анализируемого вещества необходимо соблюдать постоянство температуры.
С увеличением длины волны показатели преломления уменьшаются. В табл. 3.6.1 приведены длины волн, при которых обычно определяют показатели преломления.
Таблица 3.6.1.
Показатели преломления воды для световых волн различной длины
|
Источник света |
Цвет линий |
Обозначе ния линий |
Длина волны, нм |
Обо зна чение |
n,при T=200C |
|
Водородная трубка Натровая лампа Водородная трубка |
Красный Жёлтый Синий |
C D F |
656,3 589,3 486,1 |
n>C> n>D> n>F> |
1,3311 1,3330 1,3371 |
При измерении показателя преломления необходим источник света, дакющий излучение определённой длины волны (натровые, ртутные, водородные лампы). Табличные показатели преломления приводятся для длины волны 589нм и обозначаются n>D>>//>
Количественно дисперсию оценивают как разность показателей преломления для различных длин волн, выражение 3.6.2. Разность n>F> – n>C> называют средней дисперсией.
D = n>λ>>2 >- n>λ>>1> (3.6.2)
Показатель преломления определяют с помощью приборов, называемых рефрактометрами. В большинстве рефрактометров измерение ведётся при дневном свете или с помощью лампы накаливания. Эти приборы снабжаются компенсаторами дисперсии.
Определение показателя преломления вещества сводится обычно к измерению предельного угла преломления на границе «жидкость – стекло».
Допустим, что первая среда является жидкостью и необходимо измерить её показатель преломления - п>1>. Вторая среда представляет собой стекло призмы с показателем преломления п>2>. Вторая среда оптически более плотная, чем первая, а это значит, что п>2> > п>1> и угол преломления меньше угла падения. С увеличением угла падения увеличивается и угол преломления. Когда угол падения равен 900, луч света скользит по поверхности раздела. Если же угол падения меньше 900, то луч претерпевает преломление и попадает в зрительную трубу прибора. Этот луч называется предельным лучом, а угол преломления – предельным углом преломления. Для двух сред относительный показатель преломления может быть рассчитан по выражению, 3.6.3.
n >отн> = sinα /sinβ = n>2> /n>1> (3.6.3)
Показатель преломления призмы п>2> всегда известен, поэтому остаётся найти показатель преломления первой среды п>1> путём измерения угла преломления β.
n>1> = n>2> sinβ
В лабораторной практике наиболее часто используются рефрактометры типа Аббе и типа Пульфриха. Большее применение нашли рефрактометры типа Аббе: рефрактометр лабораторный универсальный РЛУ, рефрактометр ИРФ-22, рефрактометр лабораторный пищевой РПЛ и др. Оптические схемы и техника работы на этих приборах одинаковы, отличаются они несколько по конструкции.
Призма Амичи состоит из трёх склеенных призм с различными показателями преломления и различной дисперсией. Призмы рассчитаны так, что при прохождении через них цветных лучей только жёлтые лучи (линии D в спектре натрия) не меняют, не меняют своего направления. Устройство такого рода получило название дисперсионного компенсатора. Меняя положение призмы Амичи (или поворачивая одну призму относительно другой). Можно лучи разложенные измерительной призмой . собрать в один луч. Его направление будет таким же как и луча D , показатель преломления соответственно n>D>.
Рефрактометры типа Пульфриха более сложны в обращении и требуют специального источника света. Шкала рефрактометра градуирована в углах и нужно, производить пересчёт их на показатель преломления по специальным таблицам.
4. Электрохимические методы анализа
Электрохимические методы анализа основаны на использовании зависимости электрохимических параметров — электропроводности, сопротивления, силы тока и др. от концентрации и природы вещества, участвующего в электрохимической реакции. Электрохимические параметры при этом служат аналитическими сигналами, при условии, что они измерены достаточно точно.
Электрохимические методы анализа в практику химического анализа вошли сравнительно давно и занимают в ней важную роль. Впервые потенциометрическое титрование было проведено в 1893 г. в институте Оствальда в Лейпциге, а в 1902 г. появились труды по применению кондуктометрического титрования. А ещё в 1830 г. А.Беккерель провёл осаждение ионов свинца и марганца на положительном электроде в процессе электролиза, тем самым, положив начало электрогравиметрии.
Сейчас электрохимические методы анализа широко применяются во всех технологических процессах, научных работах и т.д., т.к. обладают рядом достоинств. Они позволяют определить концентрацию вещества в широком интервале от 1 до 1•10-9 моль/л с высокой точностью, могут проводиться дистанционно и могут быть легко автоматизированы. Обычно электрохимические методы анализа используют для прямых измерений, основанных на зависимости — “аналитический сигнал — состав”, либо для индикации конечной точки титрования в титриметрии.
Следует отметить, что по первому типу работает большинство приборов автоматического контроля. Одни приборы измеряют электропроводность раствора анализируемого продукта, которая зависит от его концентрации и изменяется пропорционально изменению последней, другие — потенциал электрода, погружённого в раствор анализируемого продукта, величина потенциала также зависит от концентрации ионов.
Методы прямой потенциометрии (ионометрии) основаны на прямом применении уравнения Нернста. Для нахождения концентрации определяемого иона по величине ЭДС цепи или потенциала соответствующего электрода.
В ионометрии сначала по стандартным растворам строят градуировочный график зависимости величины ЭДС или потенциала соответствующего электрода от концентрации определяемого иона или градуируют измерительный прибор (рН-метр, например), а затем измеряя потенциал или ЭДС анализированного раствора — находят его концентрацию. Например, широко применяют этот метод для определения рН раствора, можно использовать также для определения концентрации ионов металлов, анионов и пр.
В настоящее время прямая потенциометрия — ионометрия развивается как новая область физико-химических исследований, основной задачей ионометрии является разработка, изучение и применение широкого круга ион-селективных электродов, обратимых к большому числу катионов и анионов.
Ион-селективные электроды получают на основе самых разнообразных веществ: твёрдых и жидких ионитов, моно- и поликристаллов, хелатов и т.д.
Появление большого числа новых электродов значительно расширело инструментальную базу потенциометрического анализа, с помощью которого осуществляется контроль за ионным составом разнообразных сред.
В объёмном анализе широко применяется косвенная потенциометрия — потенциометрическое титрование, при этом цветной индикатор заменяют электродом. Конечной задачей потенциометрического титрования определяемого компонента рабочим раствором является определение точки эквивалентности по изменению потенциала электрода в эквивалентной точке.
Классификация электрохимических методов анализа.
Электрохимические методы анализа основаны на электрохимических процессах, происходящих в электрохимических системах, состоящих из электродов и электролитов, находящихся в контакте.
Эти методы, основанные на использовании электрохимических свойств анализируемых систем в зависимости от изучаемого аналитического сигнала подразделяются на несколько больших групп — кондуктометрию, потенциометрию, полярографию, электрогравиметрию и т.д.
а) кондуктометрия — метод основан на измерении электропроводности раствора анализируемого электролита, которая зависит от концентрации электролита и изменяется пропорционально изменению концентрации.
б) потенциометрия — метод основан на измерении потенциала электрода, погружённого в анализируемый раствор, величина потенциала зависит от концентрации ионов.
в) полярография — метод основан на изучении зависимости между характером поляризации рабочего электрода и концентрацией раствора, в который он помещён. Полярографию можно применять как для непосредственного определения концентрации анализируемого вещества, так и для определения конечных точек при титровании.
г) Электрогравиметрия — метод основан на выделении из раствора определяемого вещества с помощью электролиза. При этом чистый взвешенный электрод погружают в анализируемый раствор, пропускают постоянный ток, по окончании процесса электролиза электрод вновь взвешивают. По разнице взвешивания находят массу выделившегося на электроде вещества и производят расчёт.
Существуют другие методы, кроме вышеперечисленных, но все они основаны на использовании электрохимических свойств анализируемых систем.
Химические реакции, используемые в ЭХМА.
В электрохимических методах анализа могут быть использованы реакции нейтрализации, осаждения, окисления – восстановления, комплексообразования и др.
Например, в косвенной кондуктометрии (кондуктометрическое титрование) и косвенной потенциометрии (потенциометрическое титрование) очень часто используются реакции осаждения, в результате которых наблюдается изменение электропроводности в первом случае и изменение потенциала — во втором.
Рассмотрим эти случаи.
а) Кондуктометрическое титрование.
Нитрат серебра титруют хлоридом натрия:
Ag+NO>3>- + Na+Cl - → ↓AgCl + Na+NO>3>-
Ионы серебра и хлора в процессе титрования удаляются из раствора, образуя осадок, ионы натрия приходят с титрантом и остаются в растворе, заменяя ионы серебра. Такая замена ионов приводит к изменению электропроводности, и характер такого изменения определяется подвижностью ионов, участвующих в этом обмене. В данном случае более подвижные ионы серебра (λ0 = 61,9) уходят из раствора, а вместо них приходят менее подвижные ионы натрия (λ0 = 50,1 ), что естественно, ведёт к уменьшению электропроводности раствора, которая уменьшается до полного осаждения ионов серебра, а после этого электропроводность увеличивается, что фиксируется на кондуктометрической кривой в виде излома. Чем острее угол излома, тем легче установить точку эквивалентности. Острота излома зависит от характера изменения электропроводности после достижения точки эквивалентности. При подборе реагентов в кондуктометрическом титровании следует это учитывать и подбирать таким образом, чтоб реагирующие ионы отличались своими подвижностями.
 Е
Е


V

т.э.
б) Потенциометрическое титрование.
AgNO>3> + NaCl → AgCl↓ + NaNO>3>
Здесь титрование нитрата серебра хлоридом натрия проводится с применением индикаторного серебряного электрода, потенциал которого зависит от концентрации ионов серебра и будет изменяться с её изменением в процессе титрования, это отразится на кривой титрования, скачок потенциала отразит точку эквивалентности.
E>Ag/Ag>+ = E>xc> + 0,059ℓg[Ag+]
К химическим реакциям, применяемым в ЭХМА применяются почти все те требования, что и в обычном титриметрическом анализа:
скорость реакции должна быть достаточно высокой;
реакция должна протекать строго по уравнению до конца;
должны отсутствовать побочные реакции, искажающие электрические характеристики;
в потенциометрическом титровании выбор реагентов должен обеспечивать резкое изменение потенциала электрода (скачок), который зависит от разности потенциалов электрохимических систем, от величины скачка зависит точность результата;
в кондуктометрическом титровании выбор реагентов должен обеспечивать острый угол излома на кривой титрования, т.к. от этого зависит точность определения точки эквивалентности, т.е. точности результата.
4.1 Кондуктометрические методы анализа
4.1.1 Удельная и эквивалентная электропроводность. Прямая кондуктометрия
Одним из важных свойств водных растворов является их способность проводить электрический ток. Электропроводность зависит от концентрации о природы присутствующих ионов и поэтому она может быть использована для количественного определения химического состава раствора.
Хотя одно лишь измерение электропроводности не даёт возможности аналитику идентифицировать отдельные ионы, метод с успехом применяется для определения общего содержания ионов в растворе, особенно в окрашенных или мутных растворах. Метод электропроводности оказался пригодным для определения малых количеств аммиака в биологических материалах, сточных водах и др. В этом случае аммиак отгоняют из пробы и поглощают раствором борной кислоты, замеряют удельную электропроводность и сравнивают со стандартной шкалой.
Этот метод применяется для определения одного иона в присутствии минимальных количеств других.
Метод прямой кондуктрометрии основан на зависимости электропроводности от концентрации, поэтому важным этапом определения является построение градуировочного графика, используя стандартные растворы электролита.
Градуировочный график отражает зависимость электропроводности от концентрации. Определив электропроводность анализируемого раствора по градуировочному графику находят его концентрацию
Метод кондуктометрии, обладая рядом преимуществ (точность, простота) имеет ограниченное применение, если анализируемый раствор содержит примеси, т.к. электропроводность — величина аддитивная — наличие примесей изменяет её значение.




+ + φ>1> -

 φ>1>
- - +
φ>1>
- - +



 x>1>
φ>2>
x>1>
φ>2>
 Na+
Cl-
Na+
Cl-


 x>2>
+
x>2>
+
 -
-
φ>2> +
-
Методы кондуктометрии основаны на изучении зависимости между электропроводностью и концентрацией ионов в этом растворе. Электропроводность — результат электролитической диссоциации
N
 aCl
Na+
+ Cl
–
aCl
Na+
+ Cl
–
Под действием электрического тока образовавшиеся ионы принимают направленное движение.
В поле электрического тока движущиеся ионы начинают испытывать тормозящее действие со стороны молекул растворителя и со стороны противоположно заряженных ионов.
Иондипольное взаимодействие — релаксационный эффект (φ), взаимодействие противоположно заряженных ионов — электрофоретический (х). Результатом действия этих двух эффектов является сопротивление раствора прохождению электрического тока.
Растворы электролита являются проводниками П рода — перенос электричества осуществляется ионами, которые под действием электрического тока приобретают направление и, как проводники, растворы обладают сопротивлением — R.
Величина, обратная этому сопротивлению R, называется электропроводностью.
1 R — сопротивление - ом
W = —— W — электропроводность – ом -1
R
Сопротивление раствора электролита пропорционально растоянию между погружёнными в него электродами – ℓ и обратно пропорционально их площади – S
ℓ ℓ — расстояние между электродами (см)
R = ρ —— S — площадь электродов (см2)
S ρ — коэффициент пропорциональности
Если ℓ = 1 см
S = 1 cм2, то R = ρ
ρ — удельное сопротивление столба жидкости высотой 1 см и площадью 1 см2, т.е. сопротивление 1 см3.
Удельная электропроводность Н>каппа> величина, обратная удельному сопротивлению
ℓ
Н = ——
ρ
Электропроводность раствора определяется, в основном, подвижностью (скоростью) ионов электролита и количеством переносимых ими зарядов, зависит от температуры и природы растворителя.
α — степень диссоциации электролита
x = α · c · F (z>+>u>+> + z >–>u >->) с — концентрация электролита экв/л
u>+>u >-> — скорости движения ионов F — число Фарадея
z>+>z >-> — заряды катиона и аниона ( при напряжённости электрического поля 1 в/см)
Удобнее пользоваться эквивалентной электропроводностью (она отнесена к количеству вещества).
λ — эквивалентная электропроводность — электропроводность объёма раствора, помещённого между двумя параллельными электродами, расположенными на расстоянии 1 см, который содержит 1 грамм-эвивалент растворённого вещества.
с — концентрация электролита
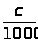 — количество г/экв в 1 см3
— количество г/экв в 1 см3
Удельная и эквивалентная электропроводность связаны соотношением:
λ =
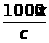
Измеряемым аналитическим сигналом в кондуктометрии является электропроводность, по мере концентрации раствора электролита — увеличивается число ионов-переносчиков электрического тока, и удельная электропроводность раствора увеличивается.
Однако, после определённого максимального значения удельная электропроводность начинает уменьшаться (у сильных электролитов увеличивается тормозящее действие со стороны молекул растворителя и со стороны противоположно заряженных ионов, а у слабых электролитов уменьшается степень их диссоциации).
Электропроводность бесконечно разбавленного раствора λ∞ определяется подвижностью ионов λ∞>-> и λ∞>+> (без учёта тормозящего эффекта — релаксационного и электрофоретического).
Кондуктометрический метод подразделяется на прямую и косвенную кондуктометрию, к косвенной кондуктометрии относится кондуктометрическое титрование, в том числе и высокочастотное.
а) Прямая кондуктометрия — метод анализа, в основе которого лежит возможность определения концентрации раствора и электропроводности, т.к. между этими величинами существует определённая зависимость.
Прямая кондуктометрия — определение электропроводности — один из методов контроля вод, грунта, пищевых продуктов и т.д.
Этот метод лежит в основе многих приборов химического контроля.
При кондуктометрическом определении газов СО>2>, СО, NH>3>, H>2>S и др. — измерению электропроводности предшествует химическая реакция. При определении СО>2> электропроводность щелочи измеряется после поглощения им СО>2>.
Используя серию стандартных растворов — строят градуировочный график зависимости (x, λ — c), затем измеряют х>х> или λх> >анализируемого раствора и по градуировочному графику определяют его концентрацию — с>х>
4.1.2 Кондуктометрическое титрование
Основано на использовании химической реакции в результате которой происходит заметное изменение электропроводности раствора, при этом могут быть использованы химические реакции всех типов — нейтрализации, осаждения, комплексообразования, окисления-восстановления и т.д.
По ходу титрования замеряют электропроводность после добавления каждой порции рабочего раствора. Зависимость электропроводности анализируемого раствора изображают графически и получают кривую кондуктометрического титрования, имеющую излом, соответствующий точке эквивалентности, по точке эквивалентности находят объём раствора, пошедший на титрование, провести расчёт.
Q>в-ва>
= Т · V>экв.
>или Q
=
 Н
Н

т.э.
 Vэкв.
V
Vэкв.
V
При кондуктометрическом титровании обязательно учитывать эффект разбавления, чтоб получить чёткий излом на кривой титрования — исследуемый раствор в электролитической ячейке должен быть разбавленным, а рабочий раствор в бюретке должен быть концентрированным, в соотношении 1 : 10.
При кондуктометрическом титровании для получения кривой титрования с резким изломом необходимо правильно подобрать рабочий раствор для титрования, растворитель для анализируемого вещества, правильно соотнести концентрации исследуемого раствора в электролитической ячейке и рабочего — в микробюретке.
Главное достоинство кондуктометрического титрования
возможность фиксировать эквивалентную точку в окрашенных и мутных растворах.
позволяет проводить анализы автоматически, дистанционно.
а) Графики кривых кондуктометрического титрования.
1. Метод нейтрализации:
а) H+Cl - + K+OH - → KCl + H>2>O
λ>H>+ = 350
λ>OH>- = 19,8
λ>Cl>- = 7,6
λ>K>+ = 63,9
Более подвижный ион Н+ замещается менее подвижным ионом К+ после т.э. — появляется избыток ОН -, более подвижный чем К+.
λ

т.э.
Vэкв. V
б) CH>3>COOH + KOH → KCH>3>COO + H>2>O
λ>H>+ = 350
λ>OH>- = 19,8
λCH>3>COO- = 40,9
λ>K>+ = 63,9
Слабая СН>3>СООН — плохо диссоциируется, образование более сильного электролита СН>3>СООН ведёт к повышению электропроводности, резко увеличивающейся при изгибе К+ и ОН –

 λ
λ



т.э. V
в) СН>3>СООН + NH>4>OH → NH>4>CH>3>COO + H>2>O
λ>H>+ = 350
λCH>3>COO- = 40,9
λNH>4>- =73,5
λ>OH>+ = 19,8
Вначале электропроводность низкая, обусловленная слабой диссоциацией СН>3>СООН, затем немного увеличивается за счёт образования сильного электролита NH>4>CH>3>COO после точки эквивалентности электропроводность остаётся постоянной, это обусловлено наличием сильного электролита NH>4>CH>3>COO, концентрация которого const.
2 Метод осаждения.
а) Ba(NO>3>) + Na>2>SO>4> → ↓BaSO>4> + 2NaNO>3>
λ>Na>+ = 50,1
λ>Ba>+ = 63,6
λNO>3>- = 71,4
λSO>4>- = 80,0
Вначале электропроводность уменьшается, вследствие замены более подвижного Ва2+ на менее подвижный Na+, после достижения т.э. — электропроводность увеличивается, за счёт появления избыточного количества сильного электролита Na>2>SO>4>/
λ


 т.э.
V
т.э.
V
б) 2AgNO>3> + BaCl>2> → 2AgCl↓ + Ba(NO>3>)>2>
λ>Ba>2+ = 63,6
λ>Ag>+ = 6,2
λNO>3>- = 71,4
λ>Cl>- = 76,0
Ионы Ag+ и Ва2+ обладают равноценной подвижностью, поэтому замещение Ag+ на Ва2+ не влияет на электропроводность, после точки эквивалентности избытка сильного электролита ВаCl>2> — вызывает увеличение электропроводности — скачок.

 λ
λ


т.э. V
При кондуктометрическом титровании после каждой добавленной порции рабочего раствора измеряют электропроводность (сопротивление).
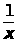 = ρ (удельная электропроводность
величина, обратная сопротивлению)
= ρ (удельная электропроводность
величина, обратная сопротивлению)
Сопротивление раствора электролита:
R
= ρ
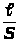 ,
отсюда x
=
•
,
отсюда x
=
•
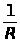
Если бы электропроводность наблюдалась только в объёме между электродами, то для определения удельной электропроводности можно было бы использовать расстояние между электродами – ℓ и их площадь – S.
Но в электролитической ячейке — ток проводит весь раствор — силовые линии находятся и между электродами и вокруг них, площадь электродов изменяется в процессе платинирования.
Поэтому удельную электропроводность выражают зависимостью:
x
= A
А — константа сосуда
(см-1),
зависящая от площади электродов и
расстояния между ними, а также от формы
сосуда и объёма раствора, проводящего
ток ( А =
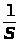 ).
).
Электропроводность раствора W — величина обратная сопротивлению R
W
=
,
и поэтому x
= A
• W
Для определения удельной электропроводности нужно измеренную электропроводность умножить на константу сосуда, т.к. константа сосуда величина постоянная — (одинаковая во всех измерениях) — то для построения кондуктометрической кривой нет необходимости пересчитывать электропроводность раствора W — в удельную электропроводность х, т.к. эти величины пропорциональны друг другу.
Основным требованием предъявляемым к электролитическим ячейкам, является постоянство константы сосуда при неизменном объёме раствора в области тех сопротивлений, которые измеряются в данной ячейке, поэтому для каждой электролитической ячейки, используемой для аналитических целей, предварительно проверяют постоянство константы сосуда.
Для определения константы сосуда измеряют сопротивление стандартного раствора с известной удельной электропроводностью, в качестве стандартных растворов применяют растворы KCl, для которых электропроводность определена с высокой точностью.
Измерение сопротивления стандартных растворов KCl 0,1 н и 0,01 н проводят при постоянном объёме раствора.
4.1.3 Порядок проведения кондуктометрического титрования
Собрать установку для кондуктометрического титрования, которая состоит из электролитической ячейки и микробюретки, установленной над сосудом для измерения электропроводности (сопротивления).
Определить константу сосуда. Для этого приготовить 0,1 н и 0,01 н растворы KCl. В начале определить сопротивление 25 мл 0,01 н раствора KCl, а затем 25 мл 0,1 н раствора KCl с помощью моста Уинстона. Константу сосуда вычисляют по уравнению:
А = х • R
где: х — удельная электропроводность раствора взятой нормальности
R — сопротивление раствора в ячейке, ом
Значения констант сосуда, установленные по 0,01 н раствору KСl должны быть близкими (6,16 и 6,07 — соответственно и практически не изменяются при увеличении объёма в ячейке).
Полумикробюретку заполнить рабочим раствором и установить над сосудом для титрования.
В электролитическую ячейку поместить 25 мл анализируемого раствора и определить сопротивление с помощью мостика Уинстона. Затем в ячейку добавить титрант порциями по 0,02 мл и после добавления каждой порции титранта раствор перемешивают и измеряют сопротивление — три раза и берут средний результат. По результатам — строят кривую титрования, по оси ординат — электропроводность раствора, отмечают точку эквивалентности и определяют объём титранта, пошедший на реакцию с определённым веществом.
 Н,
λ
Н,
λ

т.э.
V
Количество определяемого вещества находят по формуле:
Qв-ва = Т · Vэкв или
Q
=
Электропроводность (сопротивление) измеряют с помощью установки, включающей мост Уинстона.
Сопротивление раствора электролита определяют путём сравнения с эталонным сопротивлением.




 Rя
Rя

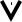 R>1>
R>1>
G
 R>2>
R>2>



С помощью скользящего контакта G подбирают такое соотношение плеч реохорда R>1> и R>2>, чтоб в диагонали моста — ток отсутствовал, тогда Rя
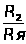 = R
Rя
— сопротивление ячейки
= R
Rя
— сопротивление ячейки
R — магазин сопротивлений
R>1> R>2> — переменные сопротивления, плечи реохорда
G — скользящий контакт
4.1.4 Установка для выполнения кондуктометрического титрования
При кондуктометрическом титровании в процессе анализа неоднократно регистрируется аналитический сигнал — электропроводность (или сопротивление), на основании этих измерений проводятся расчёты.
Для этих целей служат специальные электролитические ячейки — сосуды с вмонтированными электродами, конструкция таких ячеек должна соответствовать интервалу измеряемых сопротивлений.
Расстояние между электродами и их поверхность выбирают в зависимости от сопротивления раствора: чем выше измеряемое сопротивление, тем больше должна быть площадь электродов и меньше расстояние между ними. С учётом этого и выбирают электролитическую ячейку.
Для каждой ячейки имеется характеристика — константа сосуда, которая должна быть постоянной при неизменном объёме раствора в области тех сопротивлений, которые измеряются в данной ячейке.
Установка для кондуктометрического титрования.
2




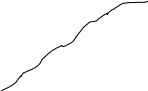


 +
+


 Rx
н.г.
Rx
н.г.






 —
4
—
4


 1
10000 3
1
10000 3


 1000
1000

 10
100
10
100
5
Расходный мост
Бюретка
Платиновые электроды
Стакан для исследуемого раствора
Магнитная мешалка
4.1.5 Порядок выполнения титрования.
В стакан для титрования помещают аликвоту исследуемого раствора и добавляют воду, чтобы электроды были полностью покрыты, м/мешалку и измеряют электропроводность, затем прибавляют рабочий раствор порциями, измеряя электропроводность после каждой порции. Следует провести 4-5 отсчётов электропроводности до точки эквивалентности и 4-5 отсчётов после точки эквивалентности. По полученным данным построить график зависимости удельной электропроводности от объёма израсходованного реактива.
 Н
Н



Т.э. V
4.2 Потенциометрические методы анализа
4.2.1 Основы метода
Потенциометрические методы анализа основаны на том, что потенциал ряда электродов является функцией активности (концентрацией), поэтому измеряя электрохимический потенциал электрода, погружаемого в анализируемый раствор — определяют концентрацию испытуемого вещества.
Потенциал электрода в растворе зависит от природы металла и от концентрации раствора, в который помещён электрод, эта зависимость выражается уравнением Нернста:
Еме.
= Е0ме.
+
 · ℓg[Me+n]
(1)
· ℓg[Me+n]
(1)
R — газовая постоянная — 8,314 Дж.
T — абсолютная температура
F — число Фарадея 96500
n — заряд иона
2,3 — коэффициент перевода ℓn → ℓg
Уравнение (1) можно представить в виде:
Еме. = Е0ме.
+
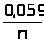 · ℓg[Men+]
· ℓg[Men+]
Е0 — нормальный, или стандартный потенциал данной окислительно-восстановительной системы, т.е. потенциал, который возникает в том случае, когда активности всех компонентов, участвующих в окислительно-восстановительном процессе, равны единице.
Окислительно-восстановительная система, определяющая потенциал электрода, может находиться в растворе, в который электрод помещают, но может возникать при погружении электрода в раствор.
В первом случае электродами являются инертные металлы (платина, золото и др.), которые не принимают участие в реакции, а служат лишь передатчиками электронов между компонентами окислительно-восстановительной системы, которые обуславливают величину потенциала электрода.
Таким примером может служить платина, погружённая в раствор, содержащий ионы Fe2+ и Fe3+ . В этом случае на электроде протекает реакция:
_
Fe3+ + e → Fe2+
Потенциал которой имеет вид:
Е = Е>0> + 0,059 ℓg[Fe3+]•[Fe2+]
Если в окислительно-восстановительной реакции участвуют ионы водорода, то потенциал электрода зависит также от величины рН раствора.
Хингидронный электрод является чувствительным на [H+]. Основной компонент электрода — эквимолекулярное соединение хинона и гидрохинона.
 О
ОН
О
ОН


 С>6>Н>4>О>2>
С>6>Н>4>(ОН)>2>—
зелёные кристаллы плохо растворимые в
воде
С>6>Н>4>О>2>
С>6>Н>4>(ОН)>2>—
зелёные кристаллы плохо растворимые в
воде
Попадая в воду, распадается на хинон и гидрохинон:
Гидрохинон диссоциирует в воде
С>6>Н>4>(ОН)>2>
С>6>Н>4>О>2>2-
+ 2Н+
_
С>6>Н>4>О>2>2-
С>6>Н>4>О>2>
+ 2е
—————————————_——
С>6>Н>4>(ОН)>2>
С>6>Н>4>О>2>
+ 2е
Происходит реакция окисления с участием [H+]
Ехэ = Е0хэ
+
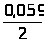 ℓg
ℓg = Е0хэ +
0,059ℓg
= Е0хэ +
0,059ℓg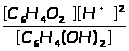
[Хиноны] и [Гидрохиноны] можно сократить, по условию они равны — смесь эквимолекулярна, отсюда:
Ехэ = Е0хэ
+
ℓg[Н+]2
= Ехэ +
• 2рН = Е0хэ
- 0,059рН
Ехэ = Е0хэ - 0,059рН
Устройство хингидронного электрода очень просто — на дне стеклянного сосуда кристаллы хингидрона и туда же опущена платиновая пластинка — подвод и отвод ē.
 Pt
Pt



 Исслед.раствор
Исслед.раствор
Кристаллы
хингидрона

Достоинство ― простота, устойчивость к загрязнению, возможность использования в неводных средах.
Недостаток ― измерения можно производить до рН не более 8 и в отсутствии окисл. и восст.
4.2.2 Электроды, применяемые в потенциометрических методах анализа
Во всех потенциометрических методах анализа используют гальванические элементы, включающие два электрода, помещённые в один и тот же раствор и измеряют ЭДС полученного гальванического элемента.
Электрод, потенциал которого зависит от концентрации (активности) определяемого иона в растворе, называется индикаторным. Индикаторные электроды могут быть электродами I и П рода.
Электроды I рода обратимы относительно ионов одного вида (металлическая пластина, опущенная в раствор собственной соли Cu/CuSO>4>, Ni/NiSO>4>).
ЕCu0/Cu2+
= E0Cu0/Cu2+
+
ℓg[Cu2+]
Часто используют электроды из серебра, ртути, кадмия, меди и некоторых других металлов. Хром, кобальт и ряд других не дают воспроизводимых результатов и электроды из этих металлов не применяются.
Для измерения окислительно-восстановительного потенциала системы применяются электроды из благородных металлов ― Pt, Au, Ir или графита.
К электродам I рода можно отнести водородный электрод, потенциал которого зависит от рН раствора.
Е = Е0
+
ℓg[H]
Е = Е0
+
рН
Наряду с электродами I рода существуют электроды П рода, потенциал которых определяется концентрацией соответствующих анионов. Такие электроды представляют собой пластину металла, покрытую труднорастворимой солью этого металла и опущенная в раствор соли, содержащей одноимённый ион.
К электродам П рода относятся хлорсеребряный, каломельный и др. Хлорсеребряный электрод изготавливают из серебряной проволоки, которую покрывают тонким слоем хлорида серебра, он помещён в раствор соли KCl.
Хлорсеребряный электрод: Ag│AgCl│KCl
EАg/Ag+
= Е>Х.С. >+
0,059ℓg[Ag+]
= E>Х.С.>
+ 0,059ℓg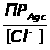
Для измерения потенциала индикаторного электрода в анализируемый раствор погружают второй электрод, потенциал которого не зависит от концентрации определяемых ионов. Этот второй электрод называют электродом сравнения.
В качестве индикаторных электродов используют электроды двух типов.
Электродообменные, на межфазных границах которого протекают реакции с участием электронов.
Ионообменные, на межфазных границах которого протекают реакции сопровождающиеся обменом ионов. Такие электроды называют ион-селективными.
А). Ион-селективные электроды.
Ион-селективные электроды широко внедряются в практику химического анализа, применяются для определения самых разнообразных веществ. Они подразделяются на несколько групп:
а) Стеклянные электроды
б) Твёрдые электроды с гомогенной или гетерогенной системой.
в) Жидкостные электроды ( на основе ионных ассоциатов, хелатов металлов, нейтральных лигандов и тд.)
г) Газовые электроды
д) Электроды для измерения активности (концентрации) биологических веществ.
Ион-селективные электроды представляют собой электрохимические полуэлементы, у которых разность потенциалов на границе раздела фаз Электрод ― Электролит зависит от концентрации (активности) определяемого иона в растворе. Электродом обычно является твёрдая или жидкая мембрана, способная обмениваться ионами с анализируемым раствором.
Если ионы в раствор электролита проникают из мембраны, то её поверхность приобретает заряд, противоположный заряду перешедших в раствор ионов и на границе раздела фаз возникает потенциал, величина которого зависит от концентрации (активности) данных ионов в растворе.
Если мембрана разделяет два раствора с различной активностью, то потенциал определяется уравнением Нернста.
Е = Е0
+ 0,059ℓg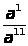
а1 ― активность (концентрация) ионов в первом растворе
а11 ― активность (концентрация) ионов во втором растворе
Обычно в одном из растворов активность (концентрацию) сохраняют постоянной (чаще внутри мембраны).
Если а11 ― const, тогда:
Е = Е0 + 0,59 ℓgа1
Т.е. потенциал индикаторного ион-селективного электрода зависит только от активности ионов первого раствора.
Ион-селективные электроды находят широкое применение в практике физико-химического анализа.
С помощью их можно быстро провести анализ по определению многих ионов, даже тех, которые другими методами не определяются.
Существуют ион-селективные электроды для определения К+, Na+, Ba2+, Ca2+, Cu2+, NO>3>-, SO>4>2-, PO>4>3-, CN -, SCN -.
Главным достоинством ион-селективных электродов является высокая избирательность определения.
Чувствительной частью ион-селективного электрода является мембрана, которая разделяет два раствора, находящихся в контакте, на внутренний и внешний, поэтому электроды называются мембранными.
Существуют различные классификации ион-селективных электродов, но наиболее удобна классификация по виду мембраны:
1 Электроды с твёрдой мембраной.
а) Стеклянные
б) С кристаллической мембраной
в) С плёночной мембраной
2. Электроды с жидкой мембраной.
3. Специальные электроды ― газовые, ферментативные и др.
В потенциометрии в качестве индикаторных обычно применяют мембранные (ион-селективные) электроды. Через мембрану возможно перемещение ионов одного вида, активность ионов внутри мембраны постоянна.
Среди ион-селективных электродов наибольшее распространение получил стеклянный электрод, предназначенный для измерения рН. Устройство его довольно простое ― включает стеклянную трубку с шариком на конце. Шарик изготовлен из специального стекла, обладающего повышенной электропроводностью и заполнен стандартным раствором ― 0,1 М раствором HCl с добавками KCl или NaCl. Токоотводом служит хлор-серебряный электрод ― серебряная проволока, покрытая хлоридом серебра, к которой припаян изолированный провод.
Шарик имеет толщину стенок 0,06-0,1 мм, изготовлен из стекла состава - 64% SiO>2>
28% Na>2>O
8% MgO
Внутри стеклянной трубки помещена серебряная проволочка, покрытая труднорастворимой солью серебра AgCl, защищенная стеклянным кожухом.
Ag, AgCl | HCl(0,1 M) || стекло || исследуемый раствор.
Перед применение стеклянного электрода для определения рН ― его вымачивают в 0,1 М растворе HCl. В результате этой операции происходит обмен ионов.
Ионы водорода из раствора кислоты обмениваются на ионы натрия в стекле шарика и на границе стекло кислота устанавливается равновесие:


Н+
Na+
В таком состоянии электрод готов к работе.
Потенциал стеклянного электрода обусловлен обменом ионов щелочных металлов, находящихся в стекле с ионами водорода раствора.
Концентрация ионов водорода на внутренней поверхности стеклянной мембраны находится в равновесии с внутренним раствором HCl и на границе мембрана ― внутренний раствор устанавливается равновесный потенциал (Е>1>).
При погружении стеклянного электрода в исследуемый раствор ионы водорода начинают перемещаться через стекло шарика (мембрану) из раствора с большой активностью, при этом на границе мембрана ― внешний раствор возникает равновесный потенциал (Е>2>).
Разность этих потенциалов даёт общий потенциал стеклянного электрода:
Ест. = Е>1> + Е>2>
Электродная реакция на стеклянном электроде сводится к обмену ионами водорода между раствором и стеклом:
Н+(р-р)
Н+(стекло)
Т.е. она не связана с переходом электронов. Ионы водорода на поверхности внешней стороны мембраны находятся в равновесии с ионами водорода в исследуемом растворе и на границе раздела возникает потенциал.
Е>1>
= Е>1>0
+
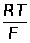 ℓn
ℓn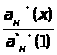
Где: а>н>+(х) ― активность ионов водорода в исследуемом растворе.
а` >н>+ (1) ― активность ионов водорода на внешней поверхности мембраны.
Аналогично на границе раздела внутренней поверхности мембраны возникает потенциал:
Е>2>
= Е>2>0
+
ℓn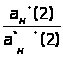
Где: а>н>+(2) ― активность ионов водорода во внутреннем растворе
а` >н>+ (2) ― активность ионов водорода на внутренней поверхности мембраны.
Суммарный потенциал стеклянного электрода:
Е>ст>
= Е>1> – Е>2>
= Е>1>0
– Е>2>0
+
ℓn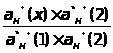
При постоянных значениях:
а` >н>+ (1) ― активность ионов водорода на внешней поверхности мембраны
а` >н>+ (2) ― активность ионов водорода на внутренней поверхности мембраны
а>н>+(2) ― активность ионов водорода во внутреннем растворе
Уравнение принимает вид:
Е>ст>
= const
+
ℓn
а>н>+(х)
Т.е. потенциал мембраны характеризует рН исследуемого раствора.
Или Е>ст> = const + 0,059ℓgС>н>+
Величина const зависит от природы вспомогательного электрода сравнения, природы внутренного раствора и др.
При определении рН, с использованием стеклянного электрода в паре с каломельным ― измеряют ЭДС цепи:
Hg, Hg>2>Cl>2>│KCl│ а>н>+(х)│стекло│HCl│AgCl, Ag
ЭДС цепи Е = Е>1> – Е>2>
Е>1> = Е0 Hg>2>Cl>2>/Hg – 0,059ℓg a>Cl>-(1) – 0,059ℓg a>н>+(х)
Е>2> = Е0 АgCl/Аg – 0,059ℓg a>Cl>-(2) – 0,059ℓg a>н>+(cт)
Е = [Е0
Hg>2>Cl>2>/Hg
- Е0 АgCl/Аg
+ 0,059ℓg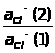 + 0,059ℓg
H+(ст)
– 0,059ℓg
aH+|(x)
= E>ст>
– 0,059ℓп а>н>+(х)
+ 0,059ℓg
H+(ст)
– 0,059ℓg
aH+|(x)
= E>ст>
– 0,059ℓп а>н>+(х)
Стеклянные электроды обладают рядом достоинств:
а) Широкий диапазон значений рН (от 0 до 13), который можно измерять стеклянным электродом.
б) Быстрота достижения равновесия и простота работы.
в) Возможность использовать электрод в присутствии окислителей, восстановителей, коллоидных растворов и пр.
Одним из недостатков стеклянного электрода является его хрупкость.
Другим недостатком ― является искажение результатов, если рН внутреннего раствора близок к рН исследуемого.
Сухие электроды очень слабо реагируют на изменение рН анализируемого раствора, поэтому перед началом измерений сухие электроды необходимо выдержать в соответствующем буферном растворе, после этого провести калибровку электрода, сверяя показания потенциала на приборе с известной рН. Выпускаемые стеклянные электроды для измерения рН (ЭСЛ - 11Г –0,5, ЭСЛ – 41Г – 0,4, ЭСЛ – 63 – 0,7, ЭСЛ – 43 – 07) пригодны для измерения рН в интервале от 0 до 14. Выпускаются стеклянные электроды для измерения активности щелочных металлов, например, ЭСNА – 51 – 7 для ионов Na+ и ЭСЛ – 91 – 07 ― для ионов К+.
К электродам с твёрдой мембраной относятся электроды с кристаллической мембраной, когда в качестве мембраны используют моно – или поликристаллы труднорастворимых в воде солей с ионным характером.


 1
― мембрана
1
― мембрана

 2
― корпус электрода
2
― корпус электрода
 6
3 ― внутренний раствор (0,1 М р-р
6
3 ― внутренний раствор (0,1 М р-р
определяемого иона и KCl)
4 ― внутренний полуэлемент Ag/AgCl
5
5 ― место припоя
4 6
― экранированный провод

 3
3
 2
2




1
Самый чувствительный участок электрода ― мембрана. Перенос заряда в кристаллической мембране происходит за счёт дефектов кристаллической решётки ― ионы перемещаются в пустующие узлы решётки.
Если мембрана неоднородна, гетерогенна ― в ней активный компонент ― кристалл внедрён в инертный связующий материал ― полиэтилен, эпоксидную смолу и т.д.
Твёрдым ион-селективным электродом является фтористый электрод, в котором монокристалл LaF>3> является мембраной, чувствительность такого электрода позволяет измерять концентрацию F – от 10 –6 до 1 м/л.
Ион-селективный электрод с мембраной из сульфида серебра для измерения ионов Ag+ и S2-. Электроды на основе сульфида серебра с добавкой соответствующего галогенида серебра позволяют измерять Cl -, J -, Br -, CN – и др. Введение в сульфид серебра сульфидов других металлов позволяет получить электрод, чувствительный к ионам металлов, внесённых со вторым сульфидом (Cd2+, Pb2+, Cu2+).
Широкое применение получают твёрдые электроды с плёночной мембраной. В таких мембранах тонкоизмельчённое активное вещество ― кристаллы ― заключено в неэлектропроводную матрицу, изготовленную из полистирола, агар-агара, каучука, полиэтилена, эпоксидной смолы и др. В качестве активного вещества применяют соли ― галогениды, сульфиты, оксалаты и др.
Конструкция электродов с плёночной мембраной аналогична конструкции электродов с кристаллической мембраной, только вместо мембраны вклеена матрица, а внутрь электрода залит раствор сравнения 0,1 м KCl и 0,1м соли измеряемого иона (для нитрат-селективного ― нитрат калия, для фторид-селективного ― фторид натрия и т.д.).
Перед работой плёночные пластифицированные электроды вымачивают в анализируемом растворе в течение суток. К электродам с плёночной мембраной относится нитрат-селективный электрод ― ЭМ – NO>3> – 01.
В настоящее время широко применяются электроды с жидкой мембраной. В электродах с жидкой мембраной раствор сравнения отделён от анализируемого тонким слоем органической жидкости, содержащей жидкий ионит, не смешивающийся с водой, но селективно реагирующий с определяемым ионом. Жидкие мембраны готовят из жидких или твёрдых ионитов или их растворов в подходящих органических растворителях, не смешивающихся с водой и могут быть катионными, анионными и нейтральными.
Существуют катионные жидкие мембраны на Са2+, Ва2+, Zn2+, Pb2+, Cu2+, Sb3+, изготовленные на основе высокомолекулярных кислот и из волей.
Анионные жидкие мембраны изготавливают на основе аминов и четвертичных аммониевых оснований.
Нейтральные жидкие мембраны могут быть изготовлены на основе органических соединений, способных связывать катионы щелочных и щелочноземельных металлов.
В качестве растворителей обычно используют эфиры, например, октиловый или дециловый эфир фосфорной кислоты, дибутилфосфат и др. в электродах этого типа возникновение потенциала на границе раздела фаз обусловлено ионным обменом, связанным с различием констант распределения между жидкой и органической фазами. Ионная селективность достигается за счёт различия в константах распределения, устойчивости комплексов и различной подвижности определяемого и мешающего ионов в фазе мембраны.
Потенциал-образующими ионами являются катионы или анионы ионных ассоциатив, т.е. электрод с катионно-анионным ассоциатом чувствителен и к катионам и к анионам, входящим в состав ассоциата. При применении мембраны для определения анионов селективность анионочувствительных электродов распределяется таким образом:
ClO>4>- > SCN - > J - > BF>4>- > NO>3>- > Br - > Cl - >J –
На основании этого ряда можно установить возможность определения одного из ионов в присутствии других. Например, открытию нитрат ионов (NO>3>-) мешают все анионы, стоящие в этом ряду влево от него и не мешают те, которые расположены вправо от него.
Устройство ион-селективного электрода с жидкостной мембраной довольно простое.
Электрод с жидкостной мембраной.
В резервуаре 2 находится ионочувствительная жидкость, органического ионита, пропитывающая мембрану. Органический ионит имеет основные, кислотные или хелатообразующие функциональные группы, растворяется в подходящем растворителе, которые не смешиваются с водой.
Для определения кальция (Са2+) в качестве жидкого ионита берут кальциевую соль алкилфосфорной кислоты RСu(O)>2>PO, растворённую в диалкилфенилфосфонате (R>2>C>6>H>5>PO) или аналогичном компоненте. В качестве раствора сравнения внутреннего серебряного электрода применяют CaCl>2>, в котором [Ca2+] постоянно и потенциал электрода будет зависеть только от концентрации иона Са2+ в анализируемом растворе. При этом с каждой стороны ион-селективной мембраны устанавливается равновесие.
СаR>2>(орг ↔) ↔ 2R(орг) + Са2+воды и Е = Е0>мембр.> 0 0,0291 ℓg[Ca2+]
Такие электроды имеют чувствительность 10 –5 – 1 м/л в области рН 6,0 до 11,0. В практике применяют ион-селективные мембранные электроды на ионы К+, Na+, NH>4>+ и некоторые другие.
Электроды с газовой мембраной позволяют определить содержание газов при анализе почвы, морской и речной воды, биологических жидкостей, промышленных газов, выхлопов и т.д.
Действие электродов основано на взаимодействии газов с водой и образованием ионов:
CO>2> + H>2>O ↔ HCO>3>- + H+
SO>2> + H>2>O ↔ HSO>3>- + H+
H>2>S + H>2>O ↔ HS - + H+
NH>3> + H>2>O ↔ NH>4>+ + OH –
Анализ сводится к определению образовавшихся в растворе ионов Н+ или ОН -, определению рН. Для работы собирают гальванический элемент, где в качестве индикаторного электрода стеклянный электрод, а в качестве электрода сравнения ― хлорсеребряный. Оба электрода помещают в жидкость с растворённым газом и определяют ЭДС элемента.
Сосуд, в котором происходит растворение газа имеет газопроницаемую мембрану, проходя через неё, газ растворяется и с помощью электродов определяется концентрация обращающихся ионов в растворе.
Ион-селективные электроды служат в качестве индикаторных и они отличаются большой чувствительностью. Предел обнаружения ионов с их помощью 10 –5 – 10 –7 м/л (иногда до 10 –19 м/л), минимальное количество пробы для одного определения 0,05 – 1 мл. Они отличаются высокой селективностью, особенно мембранные электроды.
В). Коэффициент селективности
Мембранные электроды проявляют селективность по отношению к ионам одного вида, концентрацию которых можно измерить в присутствии других ионов, не входящих в состав мембраны. Важной характеристикой ион-селективных электродов является его коэффициент селективности, который показывает, во сколько раз электрод более чувствителен к данным ионам, чем к посторонним (мешающим). Например, коэффициент селективности натриевого электрода по отношению к ионам калия составляет 1000, т.е К>с>Na+/K+ = 1000, это значит, что данный электрод в 1000 раз чувствительнее к ионам Na, и если он имеет потенциал Е при [Na+] = 10 –3моль/л, то для достижения такого же потенциала потребуется [K+] = 1 моль/л. мембранные электроды проявляют селективность по отношению к ионам одного вида, концентрацию которых можно измерять в присутствии посторонних ионов, не входящих в состав мембраны. Селективность мембраны в этом случае зависит от способности ионов мембраны обмениваться с посторонними ионами раствора. Например, если в мембране содержатся иона Са2+, а в растворе кроме них ещё и посторонние Sr2+, то селективность мембраны по отношению с Са2+ характеризуется степенью возможности обмена:
Sr2+ + Ca2+ → Sr2+ + Ca2+
Раствор мембрана мембрана раствор
К>р>
=
 >
>
Найдём К>р>, чем больше К>р>, т.е. чем больше равновесие сдвинуто вправо, тем меньше селективность.
Селективность зависит также от соотношения подвижностей Sr2+ и Са2+ в мембране и уменьшается с увеличением этого соотношения.
ŪСа2
КСа2/Sr2+
=
 ———
= К>р>
———
= К>р>
ŪSr2+
ŪСа2 и ŪSr2+ — подвижность ионов Са2+ и Sr2+ в мембране
К>р> — константа равновесия реакции обмена в мембране.
Это соотношение представляет коэффициент селективности иона Са2+ по отношению к иона Sr2+, который является количественной мерой чувствительности электрода к двум ионам. Потенциал ион-селективного электрода зависит от концентрации определяемого иона в растворе, и он всегда играет роль индикаторного электрода.
4.2.3 Прямая потенциометрия – ионометрия.
В потенциометрических методах анализа применяют гальванический элемент, состоящих из двух электродов. Один электрод является индикаторным, потенциал его зависит от концентрации (активности) определяемого иона. В качестве индикаторных электродов можно применять ион-селективные электроды, чувствительные на определяемый ион, с мембраной разного вида.
Для измерения потенциала индикаторного электрода в анализируемый раствор погружают второй электрод, потенциал которого не зависит от концентрации определяемого иона и называется электродом сравнения. В качестве электродов сравнения можно применить нормальный водородный электрод, потенциал которого равен нулю при н.у., а также электроды П рода — хлорсеребряный, каломельный и ряд других. Основным требованием к электродам сравнения является постоянство его потенциала, простота изготовления. Постоянство потенциала обеспечивается, если поддерживать постоянной концентрацию внутреннего раствора. Хлорсеребряный электрод (Ag, AgCl/KCl) чаще других применим в качестве электрода сравнения. Его можно применять в паре со стеклянным электродом при определении рН раствора, а также в паре с некоторыми ион-селективными электродами.
Электрохимическая схема пары стеклянный электрод — хлор-серебряный
Ag | AgCl | HCl(0,1) стекло||исслед. р-р || KCl | AgCl | Ag
Стеклянный электрод насыщенный хлор-серебряный
электрод
Во всех проводимых определениях с использованием методов прямой потенциометрии-ионометрии используется зависимость потенциала индикаторного электрода (обычно ион-селективного) от активности или концентрации определяемого компонента, используя для расчётов метод градуировочного графика, метод добавок, молярного свойства и т.д.
Применение ион-селективных электродов позволяет быстро решать многие аналитические задачи и даёт возможность проводить многочисленные задачи на основе составленных матриц. Например, используя ион-селективный электрод (нитрат-селективный) можно быстро и точно определить содержание нитрат-иона в технических, биологических, экологических и других объектах. (Определение нитрат-иона другими методами представляет сложную аналитическую задачу, трудоёмкую, состоящую из нескольких стадий).
Используя ионометрию, составляют гальванический элемент из нитрат-селективного пластифицированного электрода (с плёночной мембраной) и хлорсеребряного электрода сравнения.
По точной навеске методом разбавления готовят серию стандартных растворов KNO>3> (или NaNO>3>), при этом стандартные растворы готовят на фоне 1 м K>2>SO>4>, чтобы иметь постоянную ионную силу раствора. Погружают электроды в стандартные растворы (от разбавленного к концентрированному) и регистрируют зависимость ЭДС гальванического элемента от концентрации нитрат-иона, строят калибровочный график Е = f(с) или Е = f(-ℓg c). Затем берут навеску анализируемого образца на аналитических весах (до 0,0001 г) переносят в мерную колбу на 100 мл, добавляют до метки и в том же аппаратном исполнении определяют ЭДС.
По калибровочному графику находят С или -ℓg С
4.3.4 Потенциометрическое титрование
Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала индикаторного электрода. Как и в прямой потенциометрии, для потенциометрического титрования собирают цепь из индикаторного электрода, чувствительного к определяемому иону и электрода сравнения, но метод имеет ряд преимуществ:
Позволяет вести определение в присутствии посторонних веществ, влияющих на потенциал индикаторного электрода, путём подбора титранта, реагирующего с определяемым веществом.
При использовании электродноактивных титрантов позволяет определить вещества, для которых отсутствуют селективные электроды.
При окислительно-восстановительном титровании в качестве индикаторного используют электрод из Pt или другого благородного металла.
При кислотно-основном титровании в качестве индикаторного может быть использован стеклянный электрод или другой рН-чувствительный, например хингидронный.
При реакции осаждения выбирают электрод, чувствительный к определяемому веществу или к применяемому титранту. Например, серебряный электрод может быть применён как индикаторный при осаждении ионов серебра, а так же для определения ионов (Cl -, Br -, CN -, SCN -, AsO>4>3-, CrO>4>2-) образующих малорастворимые соли серебра при использовании в качестве титранта раствора AgNO>3>.
Комплексонометрическое титрование обычно проводят с металлическими электродами, соли меди — с медным, соли никеля — с никелевым, или соответствующим ион-селективным.
Задача потенциометрического титрования сводится к определению объёма титранта, затраченного на реакцию, к определению точки эквивалентности. Самый удобный и простой способ определения точки эквивалентности по кривой титрования, которая строится по результатам титрования. При этом на оси абсцисс откладывается объём прилитого раствора — (мл), а на оси ординат — соответствующее значение Е (ЭДС) ячейки.
В точке эквивалентности наблюдается резкий скачок ЭДС. В зависимости от выбранной величины кривая титрования может быть интегральной, выражающей прямую зависимость ЭДС системы от объёма прилитого раствора и точка эквивалентности находится по её перегибу (похожа на кривую титрования в методе нейтрализации).
Дифференциальная
кривая выражает зависимость изменения
величины ΔЕ : ΔV
от объёма прилитого рабочего раствора,
кривая имеет вид пика, max
пика соответствует точке эквивалентности.
Δ Е/ΔV
Е/ΔV

 Q>в-ва>
=
Q>в-ва>
=
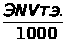
Q>в-ва> = TV>т.э.>

Vт.э. Vмл
Установка для потенциометрического титрования.






 5
5





 4
4
2
 3
3

1
1 — магнитная мешалка
2 — электролит ячейка с анализируемым раствором
3 — индикаторный электрод (ст)
4 — электрод сравнения (Х 1/с)
5 — бюретка
6 — рН метр
Расчёт кривых титрования и скачка титрования в кислотно-основном титровании.
Рассмотрим этапы титрования 100 мл 0,1 н HCl раствором 0,1 н NaOH, в качестве индикаторного используется хингидронный электрод, потенциал которого зависит от [H+]
Ехг. = Е0хг. + 0,059 ℓg[H+]
Е0хг. = 0,099; Ехг. = 0,099 – 0,059 рН; Ехг. = 0,099 + 0,059ℓg[H+]
А) к 100 мл 0,1 н HCl — 90 мл 0,1 н NaOH
[H+]
=
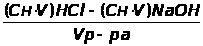
[H+]
=
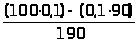 = 5,26 ·10-3
= 5,26 ·10-3
рН = -ℓg5·10-3 = 3ℓg10 - ℓg5 = 2,3
Ехг. = 0,099 – 0,059·2,3 = 0,099 – 0,135 = 0,564
Б) к 100 мл 0,1 н HCl — 99 мл 0,1 н NaOH
[H+]
=
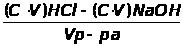 =
=
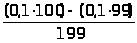 = 5·10-4
= 5·10-4
рН = -ℓg5·10-4 = 4ℓg10 - ℓg5 = 3,3
Ехг. = 0,099 – 0,59·3,3 = 0,699 –0,195 = 0,504
В) к 100 мл 0,1 н HCl — 99,9 мл 0,1 н NaOH
[H+]
=
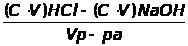 =
=
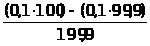 = 5·10-5
= 5·10-5
рН = -ℓg5·10-5 = 5ℓg10 - ℓg5 = 4,3
Ехг. = 0,699 – 0,059·4,3 = 0,699 – 0,254 = 0,445
Г) в точке эквивалентности рН = 7
Ехг. = 0,699 – 0,059·7 = 0,699 – 0,413 = 0,286
Потенциометрический метод позволяет вести количественное определение смеси кислот, если Kg их различаются не менее, чем на три порядка. При титровании смеси, содержащей соляную и уксусную кислоту на кривой титрования обнаруживается два скачка, первый свидетельствует об окончании титрования HCl, а второй — при оттитровывании СН>3>СООН. Несколько скачков при титровании многоосновных кислот (H>3>PO>4>, H>2>CrO>4> и др).
На основании полученных данных титрования можно построить дифференциальную кривую в координатах ΔЕ/ΔV – V, она будет иметь вид пика.
Д) после достижения Т.Э. — в избытке NaOH добавлено 100,1 мл NaOH
[OH
-] =
 =
=
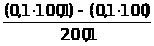 = 4,99·10-5
= 4,99·10-5
рОН = -ℓg5·10-5 = 5ℓg10 - ℓg5 = 5 – 0,7 = 4,3
Ехг. = 0,699 – 0,059·9,7 = 0,699 – 0,572 = 0,127
Скачёк титрования от недостатка 0,1 до избытка 0,1 ΔЕ = 0,445 – 0,127 = 0,318
Данные для расчёта дифференциальной кривой (метод нейтрализации).
|
Объём раствора NaOH, V мл |
ΔV |
E |
ΔE |
ΔE/ΔV |
|
0 |
0,699 |
0,015 |
||
|
9,0 |
0,135 |
|||
|
9,0 |
0,564 |
0,066 |
||
|
0,9 |
0,06 |
|||
|
9,9 |
0,504 |
0,62 |
||
|
0,09 |
0,059 |
|||
|
9,99 |
0,445 |
15,9 |
||
|
0,01 |
0,159 |
|||
|
10,0 |
0,286 |
1,59 |
||
|
0,1 |
0,159 |
|||
|
10,1 |
0,127 |
5 Хроматография
Хроматография — метод разделения и анализа смесей веществ, основанный на различном распределении их между двумя несмешивающимися фазами — подвижной и неподвижной.
При контакте с поверхностью неподвижной фазы (НФ) компоненты смеси распределяются между подвижной фазой (ПФ) и неподвижной фазой (НФ) в соответствии с их свойствами (адсорбируемостью, растворимостью или др.)
Устанавливается динамическое равновесие, вследствие чего молекулы разделяемой смеси часть времени находятся в НФ, а часть — в ПФ, а разные вещества обладают различным сродством к подвижной и неподвижной фазой, поэтому вещества, сильные взаимодействующие с НФ, будут медленнее двигаться через хроматографическую систему по сравнению с веществом, слабее с ней взаимодействующим.
Бурное развитие методов хроматографического анализа началось с работ лауреатов Нобелевской премии А.Мартина и Д.Синджа, где были предложены и разработаны методы распределительной хроматографии (1941 г.). В 1952 г. были получены первые работы в области газожидкостной хроматографии, были усовершенствованы конструкции систем ввода проб, созданы чувствительные детекторы. Метод газовой хроматографии — первый из всех хроматографических методов, получивший инструментальное обеспечение.
Начиная с 70-х годов происходит бурное развитие жидкостной хроматографии, создаются новые сорбенты и высокопроизводительное оборудование, позволяющее анализировать сложные смеси, содержащие десятки и сотни различных веществ.
В настоящее время жидкостная колоночная хроматография является одним из наиболее интенсивно развивающихся методов аналитической химии.
Хроматография. Общие принципы и классификация
Хроматографический метод основан на распределении вещества между двумя несмешивающимися фазами, одна из фаз подвижна — ПФ, а другая неподвижна — НФ. Метод можно представить как процесс многократного повторения фактов сорбции и десорбции вещества при движении его в потоке ПФ вдоль неподвижного сорбента — НФ, это наблюдается при прохождении потока газов, паров, жидкостей через колонку, содержащую зернённый слой сорбента.
Подвижной фазой является смесь, она может быть жидким раствором или газовой смесью, неподвижной фазой является сорбент твёрдый с большой поверхностью, сорбент может быть жидким, нанесённый тонкой плёнкой на поверхность твёрдого носителя.
Хроматографические методы анализа получили широкое распространение благодаря соей универсальности, экспрессивности и высокой чувствительности. Применяется широко в различных областях промышленности, науки и техники, в экологии, медицине, биологии, криминалистке и т.д.
Классификация хроматографических методов анализа
I. По агрегативному состоянию подвижной фазы:
А) Газовая хроматография — подвижная жидкость – газ.
Б) Жидкостная хроматография — подвижная фаза — жидкость.
При этом возможны следующие варианты:
|
№ п/п |
Наименование метода |
Неподвижная фаза |
Подвижная фаза |
|
1 |
Газо-адсорбционная хроматография |
Твёрдая |
Газовая |
|
2 |
Газо-жидкостная хроматография |
Жидкая на твёрдом носителе |
Газовая |
|
3 |
Жидкостная адсорбционная хроматография |
Твёрдая |
Жидкая |
|
4 |
Жидкостная распределительная хроматография |
Жидкий поглотитель на твёрдом носителе |
Жидкая |
П. По механизму разделения смеси:
а) Адсорбционная хроматография основана на различной адсорбционной способности веществ на данной адсорбенте.
б) Ионно-обменная хроматография основана на способности веществ обмениваться ионами друг с другом.
в) Осадочная хроматография основана на различной растворимости осадков.
г) Распределительная хроматография основана на различном распределении веществ (с разными коэффициентами распределения).
Ш. В зависимости от способа относительного перемещения фаз — подвижной фазы вдоль неподвижной различают следующие виды хроматографии:
1. Проявительная (элюентная) хроматография.
При работе по этому методу разделяемая смесь переносится потоком вещества (элюента), который сорбируется хуже, чем любой компонент смеси.
Через слой сорбента, находящийся в хроматографической колонке, непрерывно пропускают поток элюента, называемого носителем (он может быть газообразным или жидким). В поток носителя на входе в колонку вводят небольшой объём разделяемой смеси, содержащей компоненты А и В, которая увлекается потоком носителя и продвигается по колонке через слой сорбента.
Если компонент В сорбируется лучше, чем компонент А, то при движении смеси по колонке компонент В задерживается сорбетном сильнее и отстаёт от компонента А, и они пространственно разделяются, компонент А занимает часть объёма колонки впереди, а компонент В — часть объёма позади. Эти части объёма называются зонами компонентов, при этом компоненты находятся в зонах не в чистом виде, а в смеси с элюентом, и выходят из колонки в порядке возрастания их сорбируемости.
Выходящий из колонки поток носителя — элюента вступает в детектор, регистрирующий определённое свойство потока (например, теплоноситель).
Когда через детектор проходит зона компонента, детектор выдаёт сигнал, т.к. свойство потока изменяется и величина сигнала пропорциональна содержанию компонента в носителе.
Последовательность сигналов детектора, записанная на ленте самописца, называется хроматограммой.
Достоинствами проявительного метода хроматографии является:
При выборе подходящих условий из колонки выходят зоны всех компонентов, отделённые чистым элюетном, т.е. происходит полное разделение смеси.
Время удерживания каждого компонента при заданных условиях хроматографирования является постоянной величиной и может быть использовано для идентификации веществ.
Не требуется дополнительной регенерации сорбента, т.к. он непрерывно регенерируется элюентом.
2. Фронтальная хроматография.
Это простейший по методике вариант хроматографии, он состоит в том, что через колонку с сорбентом непрерывно пропускают смесь компонентов А, В, С, Д в растворителе S. Если сорбируемость компонентов растёт в ряду А < В < С < Д …, то на выходе из колонки сначала появляется компонент А, затем смесь А + В, затем смесь А + В + С , а потом исходная А + В + С + Д.
Хроматограмма в этом случае будет иметь вид ступенчатой кривой, т.е. детектор будет выдавать сигналы в соответствии со свойствами потока, выходящего из колонки, т.к. свойства потока изменяются, изменяется и сигнал, ему соответствующий, величина сигнала соответствует содержанию компонентов в носителе.
С игнал
детектора
игнал
детектора
А
+ В + С + Д


 А
+ В + С + Д + S
А
+ В + С + Д + S

 А
+ В + С + S
А
+ В + С + S
 А
+ В + S
А
+ В + S
 А
+ S
А
+ S

 Раств.
S
Раств.
S


Время
Фронтальный метод целесообразно применять для очистки раствора от примесей, которые сорбируются существенно лучше, чем основной компонент или для выделения из смеси наиболее слабо сорбирующихся веществ.
Таким образом, в фронтальном методе при постоянном введении в хроматографическую колонку компонентов в чистом виде можно выделить только один компонент, наиболее слабо сорбирующийся, а остальные выйдут из колонки в виде смеси.
3. Вытеснительная хроматография.
В этом методе анализируемую смесь компонентов А и В в растворителе вводят в колонку и промывают раствором вещества Д (вытеснителя), сорбируется лучше, чем компоненты А и В.
Вытеснитель Д вытесняет компонент, имеющий более высокую сорбируемость, например В, и вытесняет менее сорбируемый компонент А. Происходит перемещение компонентов А и Д вдоль слоя сорбента со скоростью, равной скорости движения вытеснителя Д.
При этом образуются зоны компонентов, вплотную примыкающие одна к другой и расположенные в порядке возрастания сорбируемости компонентов. Из колонки последовательно выходят компоненты А и В в соответствии с их избирательной сорбируемостью.
Концентрация раствора при этом методе не уменьшается, но большим недостатком является наложение зоны одного на зону другого, поскольку зоны компонентов не разделены зоной растворителя. Д



Сигнал детектора А + В


 В
В
 Д
А + В
Д
А + В
В
А А + В

 А
+ В А В
А
+ В А В



Время
АIV. В зависимости от способа оформления процесса различают колоночную хроматографию и плоскую (на бумаге и тонкослойную)
В колоночной хроматографии — неподвижная фаза в виде мелкозернистого твёрдого адсорбента или инертного материала, пропитанного определённой жидкостью, находится в длинной узкой трубке. Такая колонка называется насадочной.
Применяются также капиллярные колонки, в которых неподвижная фаза покрывает тонким слоем внутреннюю стенку колонки.
В тонкослойной хроматографии используются стеклянные пластинки, на которых нанесен тонкий слой сорбента, а также листы специальной хроматографической бумаги.
V. По назначению хроматографию подразделяют на:
аналитическую — для проведения качественного и количественного анализа
препаративную — для выделения небольших количеств чистых веществ
промышленную — для получения чистых веществ в больших количествах
5.1.2 Теоретические основы хроматографии.
Понятие о хроматограмме. Расшифровка хроматограмм
(расчёт на основе её параметров).
Задачей теории хроматографии является установление закона движения хроматографических зон и выбор оптимальных условий разделения.
Хроматографическое разделение основано на различной сорбируемости компонентов. Сорбируемость описывается изотермой сорбции, отражающей зависимость концентрации в неподвижной фазе (сорбенте) — Са от концентрации в подвижной фазе — С.
Са

С
Линейные участки изотерм, соответствующие малым концентрациям линейны и описываются законом Генри.
Са = К · С
К — константа Генри, она характеризует сорбируемость данного компонента, чем больше К, тем больше сорбируемость.
Разделение определяемого компонента между фазами.
В любой хроматографической системе происходит обратимый переход молекул определяемого компонента А из подвижной фазы (ПФ) в неподвижную (НФ), при этом:
Сп.ф. ↔ Сн.ф. (устанавливается равновесие)
Сорбируемый процесс в хроматографической колонке характеризуется константой равновесного распределения или коэффициентом равновесного распределения, который представляет собой отношение равновесной концентрации вещества в неподвижной фазе к концентрации вещества в подвижной фазе:
Краспр. = [Сн.ф.] / [Сп.ф.]
Рассмотрим Краспр. для компонента А:
Краспр. = [Ан.ф.] / [Ап.ф.]
[Ан.ф.] = mАн.ф. / Vн.ф. [Ап.ф.] = mАн.ф. / Vп.ф.
отсюда: Краспр. =
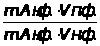 k`
=
k`
=
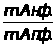 Краспр. = k`
Краспр. = k`
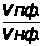
где: mАн.ф., mАп.ф. — количество компонента А в неподвижной и подвижной фазах
Vп.ф., Vн.ф. — объёмы подвижной и неподвижной фазы
K — коэффициент ёмкости
Коэффициент распределения зависит от природы определяемого компонента, природы подвижной и неподвижной фазы, температуры, рН, концентрации и др. скорость движения зоны данного вещества обратно пропорциональна коэффициенту распределения Краспр.
При больших значениях Краспр. — большая часть определяемого компонента находится в неподвижной фазе (НФ) и перемещается медленно.
Если Краспр. малая величина, то определяемый компонент быстро продвигается по колонке.
Два компонента с различными Краспр. будут перемещаться с разными скоростями, что является определяющим фактором хроматографического разделения.
При хроматографических определениях вещество подвижной фазы (ПФ) вступает в контакт с участками неподвижной фазы (НФ) — сорбента, проходит через весь его слой и на выходе из колонки поток подвижной фазы — носителя вступает в детектор, который регистрирует изменение свойств потока, изменения эти происходят за счёт изменения концентрации определяемого компонента в потоке.
Понятие о хроматограмме.
Вещество подвижной фазы, содержащее жидкий или газообразный носитель и определяемые компоненты вступает в контакт с участками неподвижной фазы — сорбента, проходит весь его слой и попадает в детектор, который регистрирует изменение свойств потока, эти изменения происходят за счёт изменения концентрации определяемого компонента в потоке. Это приводит к изменению сигналов детектора, фиксируемых лентой самописца. Эта запись, как уже упоминалось, называется хроматограммой. Хроматограмму называют также выходной кривой. Она служит выражением результатов хроматографического разделения веществ.
Сорбционная способность неподвижной фазы характеризуется временем удерживания (tR) или объёмом удерживания (VR), который представляет объём подвижной фазы, прошедшей через слой сорбента за время tR. Между ними зависимость
υ>R> = t>R> · υ υ — объёмная скорость подвижной фазы
Нулевая линия — часть хроматограммы, полученная при регистрации сигнала детектора во время выхода из колонки чистой подвижной фазы. Существуют другие параметры хроматограммы
С

 tR>2>
tR>2>

 tR>1>
tR>1>


 ΔtR>2,1>
ΔtR>2,1>
h

3
4
 tR>0>
tR>0>


 2
1
2
1

 μ
μ
>0,5>
μ
μ
>0,5>



V
1 — нулевая линия; 2 — пик несорбируемости компонента; 3, 4 — пики определяемых компонентов.
Высота выходной кривой — высота пика h — перпендикуляр из максимума пика на нулевую линию.
Ширина пика μ — отрезок отсекаемый на нулевой касательной к кривой в точке перегиба (или на половине высоты — μ>0,5>).
t>R> — промежуток времени от момента ввода пробы до достижения max h на пике.
Расшифровка хроматограммы.
Расшифровка хроматограммы сводится к определению высоты и ширины пика, если пик узкий, то определяют только высоту пика. Высота пика определяется как высота перпендикуляра, проходящего через max точку пика на нулевую точку пика, если пик пологий, то высоту проводят из точки пересечения касательных. Для широких пиков определяется не только высота, но и ширина, т.к. устанавливается зависимость между концентрацией компонента и высотой и площадью пика.





 h
h
h
h
1/2h
h


 a
a
a
a
h = fC S = fC S = h · ½ a — основание
Ширина пика находится как ширина основания треугольника, полученного при пересечении двух касательных с нулевой линией. (иногда используют 1/2h).
Хроматографический метод анализа можно использовать как для качественного, так и для количественного анализа.
1) В качественном анализе используют несколько методик расшифровки хроматограмм.
а) качественный анализ по времени удержания Rуд. (τуд.)
Rуд. (τуд.) — время удержания – это промежуток времени от момента ввода пробы до выхода max на хроматограмме, оно зависит от условий хроматографирования, от природы анализируемого компонента. Метод заключается в том, что отмечают τуд. эталонной смеси, затем исследуемой. При сравнении судят о составе смеси.
б) качественный анализ по форме хроматограмм.
Вначале получают хроматограмму исследуемой смеси, затем вводят в анализируемую смесь предполагаемое вещество.
Если введённое вещество в смеси есть, то увеличивается величина соответствующего пика, если отсутствует — появляется дополнительный пик.

 а)
б) в)
а)
б) в)


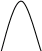
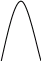
Хроматограмма в-ва нет есть
(первичная)
Если в анализируемую смесь, имеющую хроматограмму (а) ввести этиловый спирт и прохроматографировать, то может быть два новых вида хроматограмм.
(б) — говорит об отсутствии этилового спирта
(в) — говорит о присутствии этилового спирта
в) табличный качественный анализ с применением хроматографирования исследуемого и введённого эталонного раствора.
На основании полученных хроматограмм рассчитывают относительно удерживаемые объёмы по времени удержания и сравнивают с табличными данными и по ним проводят идентификацию:
V>отн.> = (τ>уд.>иссп. – τ>0>) / (τ>уд.>эт. – τ>0>)
Где: τ>уд.>иссп — время удержания исследуемой смеси
τ>уд.>эт — время удержания эталона
τ>0> — время удержания газа-носителя
Иногда используют не время удержания, а расстояние в мм (i) от момента вкалывания пробы до появления max пика.
V>отн.> = (i>уд.>иссп. – τ>0>) / (i>уд.>эт. – τ>0>)
2) Количественный анализ основан на методиках, учитывающих изменение различных параметров пика, зависящих от концентрации анализируемых компонентов — h, a, S и V>R> или hV>R>.
а) Метод нормировки — сумму параметров пиков (h, S) принимают за 100 % и массовая доля находится как отношение h и S отдельных пиков к этой сумме (х100)
% =
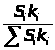 · 100 % =
· 100 % =
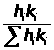 · 100
· 100
б) Метод абсолютной калибровки — наиболее точен. В нём экспериментально определяют зависимость h или S от концентрации и строят калибровочные графики по стандартным растворам, а потом хроматографируют смесь и по высоте полученных пиков определяют С.
Если тщательно готовить стандарт смеси и выдерживать условия хроматографирования, метод отличается высокой точностью.
в) Метод внутреннего стандарта основан на введении в анализируемую смесь точно известного количества стандарта, близкого по физическим свойствам к компонентам смеси. Смесь хроматографируют, определяют h и S.
% =
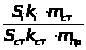 · 100
· 100
Дать пример расчёта на метод нормировки и внутреннего стандарта.
Примеры расчётов.
1. При хроматографировании смеси компонентов, расшифровка хроматограммы дала следующие данные:
|
h |
a>1/2> |
k |
S |
S>i>k>i> |
|
|
Пропан |
110 |
9 |
1,13 |
990 |
1118,7 |
|
Пентан |
71 |
10 |
1,11 |
710 |
788,1 |
|
Бутан |
22 |
7 |
1,11 |
154 |
170,94 |
Определить %-ное содержание компонентов в смеси, используя метод нормировки.
2. Вычислить % толуола в пробе по методу внутреннего стандарта, если данные хроматографирования:
(в качестве внутреннего стандарта – бензол)
3. При определении бутилового спирта методом газовой хроматографии были получены следующие пики в зависимости от содержания, используя калибровочный график:
|
С, мг |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
|
h, мм |
18 |
37 |
48 |
66 |
83 |
Для 0,02 мл исследуемого раствора получили пик h = 57 мм.
Определить %-ное содержание спирта, если ρ = 0,91 г/мл
3). Методы идентификации в газовой хроматографии.
В газовой хроматографии параметры удерживания какого-либо соединения в смеси при определённых условиях характеризуют природу этого соединения, поэтому они (параметры удерживания) могут быть использованы для целей идентификации.
В качестве параметров для идентификации чаще всего используют время удерживания t>R>, удерживаемый объём V>R>, логарифмический индекс удерживания J.
В практике качественного газохроматографического анализа используют следующие способы идентификации компонентов:
Сравнение параметров удерживания неизвестного вещества и эталонного соединения при идентичных условиях хроматографирования.
Применение графических или аналитических зависимостей между характеристиками удерживания и физико-химическими свойствами веществ (молекулярной массой, t>кип.>, числом углеродных атомов или функциональных групп и т.д.).
Сочетание газовой хроматографии с другими инструментальными методами (ИК-спектроскопией и др.).
Применение селективных детекторов.
4). Практическое применение.
Большое значение газовой хроматографии в практике вызвано тем, что с её помощью можно идентифицировать отдельные компоненты сложных газовых смесей и определить их количество. Метод является универсальным и не требует больших затрат времени.
Этим методом анализируют нефтяные и рудничные газы, воздух, продукцию основной химии и промышленности органического синтеза, нефть и продукты её переработки, производят разделение изотопов некоторых изотопов. Хроматография широко используется в биологии и медицине, в технологии переработки древесины, в лесохимии, пищевой промышленности и многих других.
Методы газовой хроматографии в физико-химических исследованиях, для анализа сложных многокомпонентных систем, определение микропримесей, а также для определения защитных свойств противогазных коробок и фильтр-поглощающих элементов.
5.1.3 Газовая хроматография (ГХ). Её виды
Подвижная фаза — газ или пар (газ-носитель).
В зависимости от состояния неподвижной фазы различают газо-адсорбционную (ГХ) и газо-жидкостную (ГЖХ) хроматографию.
В ГХ — неподвижной фазой является твёрдый адсорбент.
В ГЖХ — неподвижной фазой является жидкость, плёнка жидкости на поверхности частиц твёрдого сорбента.
Газовая хроматография основана на различной сорбируемости компонентов смеси, применима для анализа смеси газов, легколетучих жидкостей и некоторых твёрдых веществ, способных переходить в паро- или газообразное состояние.
В качестве газа-носителя используют инертные газы — Не, Ne, Ar, а также N>2>, H>2>, CO>2> и др. Скорость газа-носителя поддерживают постоянной.
Требования к газу-носителю:
Должен быть инертен по отношению к определяемым компонентам.
Должен быть химически чистым.
Быть дешёвым и легкодоступным.
Подходить к детектору.
В ГХ колонки заполняются твёрдым сорбентом. В качестве сорбентов может применяться активированный уголь, графит, силикагель, оксид алюминия, цеолиты и т.д.
Активированные угли неполярны, обладают высокой удельной поверхностью 1000-1700 м2/г, что обуславливает большую силу взаимодействия с анализируемым веществом.
Силикагель и оксид алюминия — полярные адсорбенты, на их поверхности имеются заряды.
Применяемые в качестве сорбента цеолиты, являются алюмосиликатами щелочных металлов. Их можно рассматривать как молекулярные сита, т.к. их поры имеют размеры, близкие к размерам молекул и адсорбция на них является своеобразным “просеиванием”, сорбируются, в основном, вещества, молекулы которых могут проникать внутрь кристаллической решётки.
Сравнительно недавно начали использовать полимерные сорбенты на основе сополимеров стирола, этилстирола, дивинилбензола и др.
Требования к сорбенту:
Должен быть однородным.
Должен иметь большую поверхность.
Не должен взаимодействовать ни с компонентами смеси, ни с газом-носителем.
Обладать активностью.
Не иметь каталитических свойств.
При проведении работ по методу газовой хроматографии в колонке происходит процесс адсорбции газа на твёрдом адсорбенте, при использовании газожидкостной хроматографии вместо процесса адсорбции — стал происходить процесс растворения газа в тонкой плёнке, находящейся на твёрдом носителе и эффективность разделения стала определяться не процессами адсорбции — десорбции газа, как это происходит в адсорбционной газовой хроматографии, а процессами растворения газа в жидкой плёнке и его выделения.
Дозаторы — устройства, предназначенные для ввода пробы. Проба может быть введена непосредственно в поток газа-носителя или в специальный дозирующий объём.
Небольшие количества вводят с помощью специальных микрошприцев (или медицинских). Большие по объёму пробы вводят с помощью газовых пипеток, твёрдые пробы растворяют и вводят в виде раствора с помощью микрошприца.
Принципиальная схема газового хроматографа





4




 5
5





 2
3 10
2
3 10
1
6
1. Баллон с газом-носителем Р = 100 атм.
2.
Редуктор
3.
Ротаметр, для измерения объёма газа
(расход)
4 .
Осушительная колонка
.
Осушительная колонка
5
 .
Испаритель – дозирующее устройство 8
7
.
Испаритель – дозирующее устройство 8
7
6 .
Хроматографическая колонка
.
Хроматографическая колонка
7. Детектор
8.
Регистратор - самописец
9. Конденсационные ловушки 9
10.
Термостат
Подвижная фаза в виде газа-носителя непрерывно подаётся из баллона, анализируемая проба с помощью микрошприца вводится в испаритель в поток газа-носителя и попадает в хроматографическую колонку – 6. Объём вводимой пробы от 0,0001 – 0,1 мл.
Колонка может быть прямая, V или W-образная, в форме спирали; может быть стеклянная, металлическая, пластмассовая.
Длина колонки 1-100 м ø 3-50 мм.
Для аналитических целей ℓ = 1,5-2,0 м ø 0,25-50 мм.
Чем меньше диаметр колонки, тем выше эффективность.
Металлические колонки прочнее, но плохо видно как идёт заполнение адсорбента, стеклянные — видно адсорбент, но хрупкие.
В колонке идёт основной процесс — процесс адсорбции газа на твёрдом адсорбенте, в газожидкостной хроматографии — процесс растворения газа в тонкой плёнке.
Детекторы — преобразуют информацию о составе газа выходящего из колонки в электрический или пневматический импульс. Существуют интегральные и дифференциальные детекторы.
Дифференциальные — отражают мгновенное изменение измеряемой величины, а интегральные — суммируют это значение за определённый промежуток времени.
Чаще применяют дифференциальные детекторы, основанные на применении теплопроводности газа (ДТП) или пламенно-ионизационные (ДИП).
Принцип ДТП — катарометра основан на изменении электрического сопротивления проводника в зависимости от теплопроводности. Измерительная схема моста по принципу моста сопротивления, плечи этого моста – металлические нити, сопротивление которых зависит от температуры, одна нить — в рабочей ячейке А, а вторая — в ячейке сравнения В и нагреваются постоянным током. Если через обе ячейки идёт одинаковый по составу газ, то теплоотдача одинаковая, одинаковая температура, одинаково сопротивление и сигнал равен 0, уравновешен. При изменении состава одного из потоков – характер теплоотдачи меняется, меняется температура, сопротивление и сигнал отличен от 0.
Работа ДИП основана на измерении электропроводности водородного пламени, в котором сжигают анализируемую газовую смесь. Когда горит чистый водород — ионов не образуется и электропроводность ничтожна. При сжигании пробы образуются ионы и электропроводность увеличивается.
5.1.4 Жидкостная хроматография
Среди хроматографических методов анализа наиболее разработанным является газовая хроматография, однако при некоторых анализах этот метод малоэффективен (малолетучих, химически и термически нестойких, высокореакционноспособных и др. веществ), поэтому целесообразнее применять жидкостную хроматографию.
Жидкостная хроматография основана на взаимодействии, возникающем при движении жидкой фазы сквозь неподвижный слой сорбента, обладающего большой суммарной поверхностью.
Особенностью хроматографического метода является распределение компонентов разделяемой смеси между фазами, одна из которых неподвижная большая поверхность, а другая — поток, фильтрующийся через неподвижный слой, в жидкостной хроматографии этот поток — жидкость (подвижная фаза).
Во всех случаях, когда подвижная фаза является жидкостью, мы имеем дело с жидкостной хроматографией, независимо от того, в каком состоянии находится неподвижная фаза.
Жидкостная хроматография получает всё большее развитие и применение с внедрением новых селективных адсорбентов на основе полимеров и становится высокочувствительным методом анализа многокомпонентных смесей в растворах.
В качестве хроматографической колонки в аналитической практике используют бюретки, делительные воронки.
Через колонку, заполненную адсорбентом, пропускают анализируемую смесь, состоящую из компонентов пробы и растворителя.
Эти компоненты будут распределяться на адсорбенте в зависимости от адсорбционной способности. В первую очередь (вверху колонки) будет адсорбироваться компонент с наибольшей адсорбционной способностью. Далее вниз по колонке растворяются компоненты по мере уменьшения адсорбционности. Например, имеются компоненты А и В в растворителе (Г-адсорбционная способность)
(Г) – Г>А> больше Г>В>
Для более чёткого разделения зон через колонку пропускают дополнительные порции растворителя, промывают растворителем.
А + В (растворитель)
Г>А> > Г>В>


А
 А А
А А
 А
А
В
В В В

При этом зона А смещается несколько вниз, компонент В полностью вымывается из зоны А и также смещается вниз по колонке, между зонами А и В появляется промежуток. Если промывать колонку растворителем, то можно добиться вымывания компонентов из колонки в таком порядке:
1. В 2 Растворитель 2А
Жидкость, вытекающая из колонки — элюент.
Процесс хроматографического анализа складывается из 2-х стадий:
Подготовка колонки
Получение хроматограммы
Сущность жидкостной хроматографии состоит в том, что разделяемые вещества перемещаются через слой сорбента (НФ) вместе с подвижной фазой с разной скоростью вследствие различной сорбируемости.
В классическом варианте жидкостной хроматографии через хроматографическую колонку, заполненную сорбентом (НФ) пропускают элюент (ПФ).
Элюент — жидкость движется под воздействием силы тяжести, скорость движения элюента можно регулировать.
Пробу анализируемой смеси помещают в верхнюю часть колонки, по мере продвижения пробы по колонке происходит разделение компонентов и через определённые промежутки времени отбирают фракции элюента, которые подвергают анализу с целью определения концентраций анализируемых компонентов.
Аппаратура, применяемая в классической жидкостной колоночной хроматографии постоянно модернизируется, совершенствуется.
С начала 70-х годов получила развитие современная высокоэффективная жидкостная хроматография (ВЭЖХ) — скоростная, хроматография высокого давления.
Появление этого метода было обусловлено необходимостью проведения анализа высококипящих (> 400 ºС) или неустойчивых соединений, которые не разделяются методами газовой хроматографии, а также с целью увеличения скорости и эффективности разделения.
Для осуществления этого применяют колонки с малым внутренним диаметром (2-6 мм), уменьшили диаметр частиц сорбента (5-50 мкм), что привело к необходимости увеличить давление на входе колонки до 0,5-40 Мпа. Выпускаемые промышленностью жидкостные хроматографы снабжены высокочувствительными детекторами, позволяющими определить до 10-9 – 10-10 г вещества.
Достаточно высокая скорость анализа, низкий предел обнаружения, высокая эффективность колонки, возможность определить любые вещества (кроме газов) привели к быстрому развитию высокоэффективной жидкостной хроматографии (ВЭЖХ).
В высокоэффективной жидкостной хроматографии реализуют все механизмы разделения — адсорбция, распределение, ионный обмен и др., но независимо от механизма разделения подвижной фазой (ПФ) является жидкость.
В жидкостной хроматографии применяются хроматографические параметры — t>R>, V>R> (время удерживания и удерживаемый объём).
Принципиальная схема жидкостного хроматографа.
Основные узлы
Элюент в колонку подаётся по определённой программе, состав и скорость подачи элюетна может изменяться в зависимости от условий анализа. Для обеспечения высокой скорости анализа установка жидкостной хроматографии снабжена двумя насосами (3, 4), которые управляются микропроцессором (5) и могут создавать давление до 40 МПа. Проба вводится в поток элюента через специальное устройство дозатор (инжектор) – (7). После прохождения через хроматографическую колонку (8) вещества детектируются высокочувствительным детектором (9), сигнал которого регистрируется и обрабатывается микро-ЭВМ (11). Можно зарегистрировать параметры t>R> и V>R> на ленте самописца или автоматически отобрать фракции в момент выхода пиков, чтоб провести анализ этих фракций, используя любые методы анализа.
Колонка ЖХ
Колонка в жидкостной хроматографии представляет собой трубку из нержавеющей стали с внутренним диаметром 2-6 мм и длиной 10-25 см, отполированную внутри. Колонка заполняется частицами сорбента размером 3,5-10 мкм, обычно сферической формы. Обычно используют суспензию в специально подобранном растворителе под давлением 50-80 МПа, такие колонки обладают высокой разделяющей способностью.
Детекторы
В качестве детекторов в жидкостной хроматографии обычно используют высокочувствительные спектрофотометры, которые позволяют обнаружить определяемые компоненты в количествах до 10-10 М, поглощающие свет в УФ или видимой части спектра (в пределах 190-800 нм).
В современных системах применяют высокочувствительные спектрофотометры, которые способны регистрировать спектр в течение 0,01-0,05 с, что очень важно при качественной идентификации соединений.
При анализе соединений, способных окисляться или восстанавливаться применяют электрохимический детектор, иногда применяют флуоресцентные детекторы и детекторы по электропроводности, которые чаще всего применяют в ионообменной хроматографии.
Неподвижные фазы (НФ)
Неподвижные фазы (НФ), применяемые в жидкостной хроматографии не должны смешиваться с подвижной фазой, должны быть механически и химически устойчивы в условиях анализа, обеспечивать требуемую селективность и эффективность.
В качестве таковых применяют силикагель, оксид алюминия и др.
Силикагель — (гель кремниевой кислоты SiO>2> · xH>2>O) — одна из широко используемых НФ, специфический сорбент.
Адсорбция на силикагеле происходит вследствие образования водородных связей адсорбируемого вещества с поверхностными силанольными группами ≡ Si — OH. Силикагель более прочно удерживает вещества с большим количеством водородных связей.
Для хроматографических целей используют силикагели с площадью поверхности 100-700 м2/г.
Поверхность силикагеля имеет слабо-кислый характер (рН = 3-5), поэтому соединения основного характера сорбируются на нём лучше, чем на сорбентах поверхность которых имеет основной характер (рН > 7). Силикагели применяют для разделения углеводородов, спиртов, фенолов, аминов, органических кислот, стероидов, липидов, комплексных соединений и др.
Оксид алюминия (Al>2>O>3>) — поверхность этого сорбента, образованная ионами Al3+ и О2- - способна создавать сильное электростатическое поле, обладающее поляризующим свойством, поэтому на оксиде алюминия в большей степени сорбируются легко поляризуемые соединения, имеющие систему легко смещаемых электронов, легко адсорбируется на поверхности оксида алюминия вода, которая удаляется при нагревании до 300-400ºС.
Различают три вида адсорбционных центров на оксиде алюминия:
кислотные, взаимодействующие с веществами, имеющие области с высокой электронной плотностью;
основные — адсорбирующие кислоты;
электронно-акцепторные, взаимодействующие с легко поляризуемыми ароматическими молекулами.
Количество воды на поверхности сорбента — и силикагеля и оксида алюминия влияют на процесс хроматографирования, поэтому для получения достоверных результатов хроматографирования необходимо поддерживать постоянное количество воды как на поверхности сорбента, так и в элюенте.
Кроме силикагеля и оксида алюминия в высокоэффективной жидкостной хроматографии применяют модифицированные сорбенты:
│
а) ≡ Si — O — Si — (CH>2>)>7> — CH>3> неполярный октильный силикагель
│
│
б) ≡ Si — O — Si — (CH>2>)>17> — CH>3> неполярный октадецильный силикагель
│
│
в) ≡ Si — O — Si — (CH>2>)>3> — NH>2 >полярный аминопропильный силикагель
│
│
г) ≡ Si — O — Si — (CH>2>)>3> — CN полярный цианпропильный силикагель
│

 │
│
д) ≡ Si — O — Si — (CH>2>) — неполярный фенил метиленовый силикагель
 │
│
Модифицированные сорбенты можно получить за счёт химической модификации силикагеля, силановые группы на поверхности силикагеля заменяют на различные органические соединения, что приводит к изменению селективности НФ.
│ │ │
O O O
│ │ │
Si — O — Si — O — Si —
│ │ │
O O O
│ │ │
Si — O — Si — O — Si —
│ │ │
O O O
│ │ │
(силикагель)
В качестве полярных модифицированных сорбентов используют силикагели с привитыми цианопропильными, аминопропильными, оксипропильными группами и др. (в, г).
На модифицированных полярных сорбентах значительно быстрее устанавливается равновесие при переходе от элюента, меньше погрешности анализа.
В качестве неполярных модифицированных сорбентов используют силикагели с привитыми этильными (С>2>), октильными (С>8>), октадецильными (С>18>) и фенильными радикалами. Эти сорбенты имеют большое сродство к гидрофобным молекулам (а, б, д).
5.1.4 Распределительная хроматография
Распределительная хроматография является разновидностью жидкостной и основана на распределении вещества между двумя несмешивающимися фазами.
В качестве основного показателя в этом методе является коэффициент равновесного распределения, вернее различие в величинах коэффициентов распределения отдельных компонентов раствора между двумя ненасыщенными растворителями.
При распределительной хроматографии носитель пропитывается одним из растворителей — “неподвижный” растворитель (например, водой пропитывают бумагу или силикагель). Другой растворитель (например, хлороформ) является “подвижным” растворителем и его пропускают через колонку носителя.
В качестве неподвижного растворителя чаще всего используют полярные жидкости — воду, H>2>SO>4>, CH>3>OH и др., в качестве подвижного растворителя применяют менее полярные жидкости, органические растворители.
Анализ может проводиться как в колонке, так и на бумаге и на пластинке — в тонком слое.
При обработке носителя неподвижным растворителем — на поверхности носителя образуется тонкая жидкая плёнка, колонка считается подготовленной к работе. После этого через колонку начинают пропускать исследуемую смесь в подвижном растворителе.
Подвижный и неподвижный растворители подбирают в зависимости от природы носителя.
Если носитель гидрофильный, то неподвижным растворителем должна быть вода, а подвижным растворителем — органический малоподвижный растворитель.
Если носитель гидрофобный, то неподвижным растворителем должно быть органическое вещество, а подвижным растворителем — вода. (Полярный растворитель всегда вытесняет менее полярный из полярных сорбентов).
Порцию исследуемой смеси, растворённой в подвижном растворителе, вводят в колонку и после того, как она впитывается верхней частью колонки, начинают промывание колонки подвижным растворителем.
В процессе промывания происходит непрерывное перераспределение веществ смеси между двумя несмешивающимися жидкими фазами.
Так как разные компоненты смеси имеют различные коэффициенты распределения, то и скорость передвижения компонентов по колонке различна. Наибольшей скоростью продвижения по колонке будет обладать тот компонент смеси, который имеет наибольший коэффициент распределения.
Краспр. =

т.е. отношение концентрации растворённого вещества в подвижной фазе (г/л) к его концентрации в неподвижной фазе (г/л). если коэффициенты распределения отдельных компонентов смеси достаточно между собой различаются, то при промывании колонки образуются отдельные зоны чистых веществ, т.е. происходит полное разделение смеси. (Чем больше Краспр., тем быстрее вещество движется по колонке).
Например, смесь веществ А и В пропускают через воронку в подвижном растворителе, причём Краспр. компонента В больше Краспр. компонента В, поэтому скорость распределения компонента А будет больше и компонент А уходит вниз колонки.
В


 В
В
 А
Первичная
А
Первичная
 позиция
А
позиция
А
Промывая колонку растворителем между компонентами образуется чёткая граница.
При дальнейшем промывании из колонки будет вытекать:
Подв.раств. + А
Подв.раств.
Подв.раств. + В
Разновидностью распределительной хроматографии является бумажная и тонкослойная.
В тонкослойной — разделение проводится на пластинках, покрытых тонким слоем окиси алюминия, силикагеля или другого сорбента, который удерживает неподвижный растворитель. Нижний край пластинки с нанесённой на неё пробой опускают в подвижный растворитель.
Хроматография на бумаге является разновидностью метода распределительной хроматографии, носителем для неподвижного растворителя служит при этом фильтровальная бумага, а не колонка с сорбентом. Разделение веществ происходит вследствие различия в распределении между двумя жидкими фазами, одна из которых подвижна (смесь органических растворителей), а другая — неподвижная и представляет собой воду, находящуюся в волокнах фильтровальной бумаги.
Разделение смесей веществ или ионов с помощью хроматографии на бумаге основано на различной скорости движения компонентов, которые характеризуются коэффициентом движения R>f>.
Величины коэффициентов ионов вычисляют по формуле:
R>f>
=
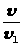 =
=
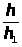 где: υ — скорость движения зоны иона на
бумаге
где: υ — скорость движения зоны иона на
бумаге
h — расстояние, пройденное зоной иона на бумаге
υ>1> — скорость движения фронта подвижного растворителя
h>1> — расстояние, пройденное растворителем
Под фронтом растворителя понимают видимую границу распространения растворителя по бумаге.
Коэффициент движения каждого катиона — постоянная величина, не зависит от концентрации анализируемого раствора, температуры, присутствия других катионов и природы аниона, с которым связан изучаемый катион.
Однако, коэффициент движения R>f> зависит от состава и свойств используемого подвижного растворителя, а также от сорта хроматографической бумаги.
Чем больше величина R>f>, тем быстрее и дальше продвигается катион по бумаге и тем лучше отделяется он от другого катиона с низким значением R>f>.
Например, у катионов Fe3+ и Cu2+ коэффициенты движения значительно отличаются по величине, поэтому они чётко разделяются.
Просто и удобно проводить разделение ионов с помощью круговых хроматограмм, полученных на обеззоленных фильтрах “синяя лента” с использованием в качестве хроматографической камеры-эксикатора. Для контакта с растворителем из фильтра вырезается полоска — “фитиль”, который погружается в растворитель (а), или контакт с растворителем осуществляется с помощью конуса, сделанного из этой же фильтровальной бумаги и вставленного в середину отверстия, находящегося в середине фильтра-хроматограммы.



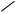 Конус
бумажный
Конус
бумажный
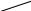 фитиль
фитиль
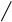
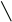

 растворитель
растворитель
 растворитель
растворитель
Для расчёта R>f> какого-то иона, например, Fе3+, на середине фильтра помещают 0,05 мл раствора, содержащего Fe3+, при этом этот процесс должен быть медленным, постепенным, чтоб происходило впитывание раствора за счёт капиллярных сил бумаги.
Образовавшееся пятно осторожно обводят простым карандашом, фиксируя его положение на бумаге, фильтр сушится и бритвой вырезается фитиль. После этого фильтр устанавливается над сосудом с растворителем, таким образом, чтоб контакт фильтра с ним осуществлялся через фитиль:



фильтр
фитиль

растворитель
в тигле
Оставляют систему в таком виде на 2-3 часа, поместив её в эксикатор с крышкой (хроматографическую камеру) для размывания первичной хроматограммы, после этого вынимают фильтр из эксикатора и отмечают карандашом границы фронта растворителя, таким образом, получаем величины:
h — расстояние, пройденное зоной иона на бумаге (для Fe3+ - экспериментально, 1,5 см)
h>1> — расстояние, пройденное растворителем (экспериментально, для смеси 90 % С>2>Н>5>ОН и 10 % 5 н HCl, 4,8 см)
отсюда RFe3+
=
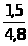 = 0,31
= 0,31
Аналогично можно определить для любого иона (Rcu2+ = 0,45).
После этого можно провести хроматографическое разделение на бумаге смеси этих катионов. Причём, учитывая величины R>f> , можно сделать заключение, что Cu2+ будет быстрее и дальше продвигаться по бумаге, т.к. имеет большую величину R>f>, чем Fe3+.
Кроме круговых хроматограмм можно использовать бумажные полосы, помещённые в стеклянные камеры, эксикаторы, пробирки. Конец этих полос помещают в растворитель (восходящая хроматография, нисходящая хроматография).
а) Восходящая бумажная хроматография.
Исследуемое вещество наносят на линию старта, которая проводится на расстоянии 1-2 см от нижнего края ХР-бумаги.
Бумагу помещают в камеру с подвижным растворителем таким образом, чтобы линия старта была выше границы растворителя. (Камера закрыта крышкой). Под действием капиллярных сил подвижный растворитель поднимается вверх, захватывая компоненты анализируемой смеси. Так как компоненты обладают различной растворимостью в этом растворителе, то и двигаться они будут с различной скоростью. При этом происходит разделение смеси на компоненты.
Если смесь А и В (К>А> > К>В>), то В будет выше В.
Линия, до которой доходит подвижный растворитель — называют линий фронта.
R>f>
=
h —
путь, пройденный растворителем от линии
старта до линии фронта
h>1> — путь, пройденный веществом от линии старта до середины пятна.
б) Нисходящая бумажная хроматография.
В этом методе растворитель находится в верхней части камеры, линия старта находится в верхней части камеры, линия старта сверху. Растворитель движется вниз под действием силы тяжести, забирая с собой определяемое вещество, чем больше Кр вещества, тем ниже на бумаге будет находиться вещество.
Распределительная хроматография на бумаге, особенно с применением органических реактивов, является макроаналитическим методом, широко применимым в тех случаях, когда обычные химические методы малопригодны.
Например, для разделения близких по свойствам соединений — аминокислот, пептидов, углеводов и т.д.
Хроматографию на бумаге используют для определения следов ФОВ в пищевых продуктах и на биологическом материале.
Ионообменная хроматография
Ионообменная хроматография основана на явлении обмена ионов находящихся в растворе и ионов, адсорбируемых твёрдым адсорбентом. Образование хроматограмм в этом случае происходит вследствие неодинаковой способности к обмену различных ионов хроматографируемого раствора. В ионообменной хроматографии, также как и в адсорбционной, можно применять фронтальный, вытеснительный и проявительный (элюентный) метод анализа.
При фронтальном анализе исследуемую смесь непрерывно подают в верхнюю часть колонки и следят за появлением отдельных компонентов в вытекающем растворе, но полного разделения компонентов в этом методе не происходит и метод не пригоден для препаративного разделения и количественного определения веществ.
При вытеснительном методе анализа для вытеснения применяют растворы веществ, ионы которых сорбируются лучше, чем ионы компонентов хроматографической смеси, поэтому они вытесняют из сорбента ранее сорбированные ионы разделяемых веществ.
В проявительном (элюентном) методе промывание проводят чистым растворителем. Во всех видах ионообменной хроматографии имеет место многократное повторение процессов ионного обмена.
В зависимости от того происходит ли обменная сорбция положительно заряженных ионов (катионов) или отрицательно заряженных ионов (анионов) — ионообменники делятся на катиониты и аниониты.
Существуют иониты, обладающие амфотерными свойствами..
а) Катионный обмен: RH + NaCl ↔ RNa + HCl
б) Анионный обмен: ROH + NaCl → RCl + NaOH
R — радикал, образующий элементарную ячейку ионита.
Качественная характеристика ионообмена зависит от природы ионита, хроматографируемого иона, растворителя, от условий опыта (tº, рН и др.).
Рассмотрим ионообменное равновесие на примере обмена на ионите двух одновалентных ионов А+ и В+.
RA + B+ ↔ RB + A+
Согласно закона действия
масс: Кр. =
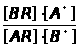 или
или
 = Кр.
= Кр.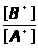
Или
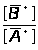 = Кр.
, где Кр. — константа ионного обмена
= Кр.
, где Кр. — константа ионного обмена
[B+], [A+] — концентрации ионов А и В в растворе
_ _
[B+], [A+] — концентрация ионов В+ и А+ в твёрдой фазе
Аналогично для обмена двухвалентного иона на одновалентный
В2+ + 2RA → R>2>B + 2A+
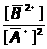 = Кр.
= Кр.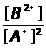
Константа равновесия (ионного обмена) позволяет количественно характеризовать сравнительную способность ионита к обмену.
Если Кравн. < 1 ион, находящийся в растворе, имеет большое сродство к иониту, чем ион, пришедший в раствор с твёрдой фазой — на ионите. Обмен из раствора в этом случае будет протекать достаточно полно.
Если Кравн. > 1 ион раствора имеет меньшее сродство к иониту, чем ион, входящий в состав ионита, обмен в данном случае незначительный.
в) Сорбенты, применяемые в ионообменной хроматографии.
Вещества, применяемые в качестве ионообменных сорбентов, подразделяются на два основных класса: неорганические и органические сорбенты, которые могут быть естественного и искусственного происхождения.
Ионообменные сорбенты должны отвечать следующим требованиям:
Обладать максимально возможной поглотительной способностью;
Обладать избирательной сорбцией по отношению к веществам разделяемой смеси;
Быть однородными, иметь степень дисперсности, достаточную для обеспечения необходимой скорости адсорбции и равновесного прохождения раствора через колонку с требуемой скоростью;
Иметь ограниченную набухаемость, не растворяться в хроматографируемом растворе и той среде, в которой они используются, обладать механической прочностью;
Производство сорбентов должно быть экономически выгодным и основываться на применении отечественного сырья.
г) Минеральные иониты.
Сорбенты минерального происхождения — слабокислотные китиониты или слабоосновные аниониты.
Наиболее распространены — оксид алюминия, природные алюмосиликаты, фосфат циркония, применяемые как катиониты.
В качестве анионитов — оксид Al, гидроксид Zr и др.
Органические иониты — являются продуктами химической переработки угля и лигнина, их называют ионообменными смолами.
д) Органические иониты.
Катиониты содержат в своих формулах сульфогруппы — SO>3>H, фосфогруппы РО(ОН)>2>, карбоксильные — СООН — (фенолформальдегидные, полистирольные катиониты).
Органические аниониты — полиамины, т.к. содержат NH>2>R+, NHR>2>+ и т.д. Аминоформальдегидные, полиаминовые, полистирольные аниониты.
Использованные иониты можно вернуть в исходную форму, пропуская через него соответствующий раствор, этот процесс называется регенерацией.
Оксид алюминия проявляет амфотерные свойства и может играть роль как катионита, так и анионита.
Al>2>O>3> — катионит Al + NaOH + CO>2> → Al>2>O>3>
На поверхности Al>2>O>3> адсорбируется NaAlO>2>, образуя соединение (Al>2>O>3>)>m>NaAlO>2>, появляются подвижные ионы Na+
[(Al>2>O>3>)>m>NaAlO>2>-]Na+ + Me+An → [(Al>2>O>3>)>m>AlO>2>-]Me + NaAn

AlO>2>- Na AlO>2>
Или (Al>2>O>3>)>m> + MeAn>2> → (Al>2>O>3>)>m> Me + 2NaAn
AlO>2>- Na AlO>2>
Al>2>O>3> — анион (перед использованием промывают азотной кислотой, в результате чего на поверхности Al>2>O>3> появляется подвижный анион — NO>3>).
[(Al>2>O>3>)>m>AlO>2>]Na + 2HNO>3> → [(Al>2>O>3>)>m>AlO+]NO>3> + NaNO>3> + H>2>O
[(Al>2>O>3>)>m>AlO+]NO>3> + MeAn → [Al>2>O>3>)>n>AlO+]An + MeNO>3>
По способности поглощать ионы для каждого ионита существуют адсорбционные ряды по способности замещать друг друга (для Al>2>O>3>):
H+ > As3+ > Sb3+ > Bi3+ = Fe2+ + Hg2+ = UO>2> > Pb2+ > Cu2+ > Zn2+ > Co2+ = Ni2+ = Cd2+ > Mn2+
Т.е. на Al>2>O>3> легче адсорбируется Н+, хуже Mn2+.
Реакции на органическом ионите (ионнообменной смоле) происходят по схеме:
2R – SO>3>H + CuSO>4> → (RSO>3>)>2>Cu + H>2>SO>4>
Основные качества ионита определяются сорбционной ёмкостью, физическими свойствами и химической стойкостью.
Ёмкость сорбента условно характеризуется количеством растворённого электролита, поглощённым единицей веса или объёма сорбента.
К показателям физических свойств ионита следует отнести величину насыпаемого веса, влагостойкость и механическую прочность.
Химическая стойкость сорбента определяется по тем рабочим средам, в которых должна проходить сорбция и регенерация.
Мерой стойкости служит степень потери веса сорбента и степени потери ёмкости.
Многие сорбенты постепенно окисляются под влиянием кислорода воздуха, теряют прочность, снижается содержание активных групп, появляются водорастворимые фракции.
е) Получение хроматограммы на колонке и её анализ.
Раствор анализируемой смеси пропускают через хроматографическую колонку.
Вследствие различной адсорбируемости компонентов смеси в колонке образуются зоны. Однако, полного разделения смеси при этом не произойдёт, только первая, самая нижняя зона будет содержать в чистом виде один наименее адсорбируемый компонент, вторая — будет состоять из смеси двух компонентов, третья — из смеси трёх и т.д.
Таким образом, при получении первичной хроматограммы можно получить в чистом виде лишь одно из смеси веществ.
Для полного разделения компонентов смеси используют операцию вытеснения (элюирования). Она заключается в том, что после получения первичной или промытой хроматограммы, через колонку пропускают растворитель, способный вытеснять все или некоторые адсорбированные компоненты смеси. При этом компоненты смеси, вытесняя друг друга, располагаются в колонке в виде отдельных чистых зон в соответствии с их способностью к адсорбируемости, а продолжительным пропусканием раствора через колонку достигают последовательного вытеснения из колонки компонентов смеси.
Например, пропуская через колонку с анионитом солянокислый раствор ионов Fe3+, Cu2+, Zn2+, Pb2+ и Bi3+ получают первичную хроматограмму этих ионов, сорбируемых на анионите.
Затем последовательно промывают колонку 2,5 н и 0,02 н HCl, а затем 2 н H>2>SO>4>.
При этом:
а) 2,5 н HCl разрушает менее прочные солянокислые комплексы Fe3+ и Cu2+ и они уходят в фильтрат;
б) 0,02 н HCl разрушает солянокислые комплексы цинка и свинца, вытесняя их из колонки;
в) 2 н H>2>SO>4> разрушает комплекс висмута и также вытесняет его из колонки.
Собирая отдельные фракции вытеснения, проводят определение компонентов в них любым физико-химическим методом — спектрометрическим, полярографическим и т.п.
Для выбора условий проведения хроматографического анализа, т.е. для изучения сорбции и десорбции каждого элемента в отдельности, строят выходные кривые. Полное разделение компонентов возможно, если между пиками концентраций имеет место чистая зона растворителя.
На приведённой кривой чёткое разделение между Cu2+, Pb2+, Bi3+, железо отделяется хуже.
Заключительной стадией хроматографического анализа смеси веществ является качественный и количественный анализ полученной хроматограммы.
Хроматограмма, полученная на адсорбенте белого цвета, представляет собой серию цветных зон, расположенных в определённом порядке.
Выявление бесцветных зон осуществляется методом проявления хроматограммы, обрабатывая зоны соответствующими реактивами или индикаторами.
Количественный анализ проводится лишь в том случае, когда осуществлено полное разделение смеси и хроматограмма состоит из отдельных неперекрывающихся зон, анализ сводится к определению вещества в каждой зоне. Широко применяется способ химического и радиохимического анализа отдельных зон, вырезанных из колонок.
При фронтальном анализе собирают последовательно порции фильтрата в отдельные приёмники и подвергают их количественному анализу химическими и физико-химическими методами.
Последовательность их распределения на колонке будет соответствовать величинам их ПР:
в верхней зоне расположится жёлтый осадок иодида серебра (ПРAgJ = 1,1·10-16);
в средней части — голубовато-серый бромида серебра (ПРAgBr = 6·10-13);
в нижней части белый осадок хлорида серебра (ПРAgCl = 1,8·10-10).
В качестве носителя используют чистые вещества, обладающие хорошей фильтрующей способностью (Al>2>O>3>, силикагель) — высокодисперсные, (BaSO>4>) — гидродисперсные.
Носитель может быть индиферентным по отношению к осадителю, хроматографируемым веществам, к образующимся осадкам.
Желательно, чтобы носитель имел светлую окраску.
Осадитель — это реагент, образующий с определяемыми ионами труднорастворимые осадки.
Осадитель должен быть индиферентным к носителю и хорошо адсорбироваться на носителе.
Осадочная хроматография может проводиться на колонке, на бумаге и на тонком слое.