Цитогенетическая и молекулярно-цитогенетическая характеристика микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи
Министерство образования и науки Украины
Харьковский национальный университет им. В.Н. Каразина
Биологический факультет
Кафедра генетики и цитологии
Курсовая работа
по генетике на тему:
Цитогенетическая и молекулярно-цитогенетическая характеристика микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи
Харьков 2010
СОДЕРЖАНИЕ
Введение
Глава 1. Современные методы и проблемы диагностики наследственной патологии
1.1 Современные представления о наследственных заболеваниях
1.2 Геномный импринтинг
1.3 Болезни импринтинга4
Глава 2. Характеристика синдромов Прадера-Вилли и Ангельмана
2.1 Цитогенетическая характеристика синдромов Прадера-Вилли и Ангельмана
2.2 Клинические проявления и методы диагностики синдромов Прадера-Вилли и Ангельмана
2.2.1 Клинические проявления синдрома Прадера-Вилли
2.2.2 Клинические проявления синдрома Ангельмана
2.3 Связь синдромов Прадера-Вилли и Ангельмана
Глава 3. Синдром Ди Джорджи (Ди Георга)
Выводы
Список использованной литературы
Приложение
ВВЕДЕНИЕ
Одной из наиболее актуальных проблем современной медицинской генетики является определение этиологии и патогенеза наследственных заболеваний. Цитогенетические и молекулярные исследования имеют высокую диагностическую информативность и ценность при решении данной проблемы, так как хромосомные аномалии встречаются с частотой от 4 до 34% при различных наследственных синдромах. В последние годы появились новые методы цитогенетических и молекулярно-цитогенетических исследований, значительно расширяющие диагностические возможности при заболеваниях, сопровождающихся различными хромосомными нарушениями. Данный обзор посвящен вопросам выбора необходимых цитогенетических или молекулярно-генетических анализов при различных формах генетических синдромов.
В работе рассматриваются генетические заболевания, вызванные микроделециями хромосом. На примере наследственных заболеваний (синдромах Прадера-Вилли, Ангельмана, Ди Джорджи) будет изучено явление геномного импринтинга, влияние которого ученые изучают и сейчас, так как до конца механизм геномного импринтинга пока не известен. Будут рассмотрены возможные варианты медицинской помощи при данных заболеваниях и риски дальнейших проявлений этих заболеваний в семьях, где встречаются такие патологии. Изучение этой проблемы на сегодня является актуальной.
Целью работы является: изучение цитогенетических и клинических проявлений микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи.
ГЛАВА 1. СОВРЕМЕННЫЕ МЕТОДЫ И ПРОБЛЕМЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
1.1 Современные представления о наследственных заболеваниях
Одной из наиболее актуальных проблем современного здравоохранения является организация медико-генетической помощи семьям, где встречаются случаи рождения детей с наследственными заболеваниями. Актуальность этой проблемы определяется широким распространением данной патологии, трудностью дифференциальной диагностики большого количества наследственных болезней, проявляющихся множественными аномалиями развития с высоким риском повторения заболевания в семье.
По данным, опубликованным в начале 90-х годов прошлого века, в мире насчитывалось около 7,5 млн. человек с наследственными патологиями, одним из клинических проявлений которых является умственная отсталость. Цитогенетические методы исследования играют огромную роль в выяснении этиологии генетических заболеваний. Согласно последним сообщениям, хромосомные аномалии среди пациентов с задержкой развития и умственной отсталостью встречаются в среднем с частотой от 4 до 34% [1, 3].
В настоящий момент появились новые методы цитогенетического и молекулярно-цитогенетического исследований, значительно расширяющие спектр современных диагностических возможностей. К этим методам можно отнести FISH-метод, основанный на гибридизации флюоресцентно меченных ДНК-зондов на разные участки генома; сравнительную геномную гибридизацию; метод спектрального кариотипирования; праймерное исследование in situ и др. В связи с разнообразием современных цитогенетических методов в литературе широко обсуждается вопрос о выборе необходимого цитогенетического или молекулярно-генетического анализа для установления диагноза при задержках психического развития и умственной отсталости [4].
В соответствии с рекомендациями согласительной конференции американской коллегии медицинской генетики (The Consensus Conference of the American College of Medical Genetics, 1997), любому пациенту с такой патологией на первом этапе необходимо провести стандартный цитогенетический анализ c разрешением в 500 сегментов. В случае подозрения на синдром, обусловленный микроструктурными аномалиями, необходимо проведение более чувствительных молекулярно-цитогенетических исследований. Первые микроструктурные нарушения хромосом, ассоциированные с определенным синдромом, были обнаружены в 1980 г. Е. Buhler и соавторами. Они сообщили о терминальной делеции сегмента 8q24 у девочки с клиническими признаками синдрома Лангера-Гидеона. К настоящему времени насчитывается около двух десятков микроделеционных синдромов, обусловленных терминальными и интерстициальными делециями разных хромосом (Рубинштейна-Тейби, Миллера-Дикера, Вильямса, Лангера-Гидеона, Прадера-Вилли, Ангельмана, Аладжилла, Ди Джорджи, Рассела-Сильвера и др.) [5, 9-12].
Микроделеции и микродупликации обычно затрагивают ряд близко сцепленных генов, доза которых в результате такой перестройки существенно меняется. В 1986 г. R. Schmickel предложил обозначать болезни, обусловленные изменением дозы близко расположенных генов в результате микроделеций или микродупликаций, термином «синдром смежных генов» (contiguous gene syndrome). Очень важно, что идентифицируются данные синдромы еще до цитологической локализации, и это в свою очередь является главным критерием их обозначения как «синдрома смежных генов». В литературе описаны случаи таких заболеваний, обусловленные не только делециями и дупликациями хромосомных сегментов, но и без них. Указанные синдромы являются преимущественно спорадическими. Отмечено, что размер вовлеченного в хромосомную перестройку района связан с тяжестью заболевания [7].
Многочисленные сообщения последних лет свидетельствуют о существенной роли субтеломерных перестроек в генезе недифференцированной умственной отсталости. Показано, что субтеломерные регионы хромосом насыщены генами, и мутации в них могут приводить к генетическим нарушениям. В настоящий момент анализ субтеломерных перестроек, проводимый различными методами, был выполнен во многих выборках пациентов с наследственными заболеваниями, у которых было выражено отставание в развитии. Фактически субмикроскопические субтеломерные хромосомные аномалии были обнаружены у 6,5-7,4% детей с умеренной и тяжелой умственной отсталостью [16, 17] и у 10,3% детей с легкой умственной отсталостью. Благодаря этим исследованиям были описаны следующие формы патологии:
- субмикроскопическая терминальная делеция 8pter, связанная с транслокацией t(8;20), приводящая к психическим и поведенческим проблемам;
- терминальная делеция хромосомы 5р у пациента с фенотипическими проявлениями синдрома Lujan-Fryns;
- тандемная транслокация 22/15 с делецией района 22q13.3 и сохранением района ядрышкового организатора хромосомы у пациента с задержкой психомоторного и речевого развития и гипотонией, без каких-либо дисморфологических особенностей;
- делеция района 22q13 у пациентов с задержкой психомоторного развития и речи, гипотонией и незначительными малыми аномалиями;
- делеция 16р, возникшая de novo, сочетающаяся с гипотонией и неспецифическими аномалиями; - субтеломерная делеция вследствие семейной сбалансированной транслокации t(3;16) (q29; р13.3), сегрегирующей в двух поколениях;
- делеция района 1р36.3 (выполнен комплексный геномный анализ карт сцепления) [2, 4]. Данная хромосомная аномалия может быть связана с гипотонией, аномалиями роста, характерным лицевым фенотипом (выпуклый лоб, глубоко посаженные глаза, плоская переносица, гипоплазия средней трети лица и выступающий подбородок), кардиомиопатией, расширением желудочков мозга, гипоплазией мозолистого тела, лейкодистрофией, психическими расстройствами [4].
Данные случаи еще раз указывают на важность выявления субтеломерных микроперестроек при спорадических и семейных формах генетических заболеваний. В связи с техническими сложностями и высокой стоимостью исследования субтеломерных участков предварительно необходим тщательный клинический отбор обследуемых пациентов. С этой целью B. de Vries и соавторы обследовали 29 пациентов с уже известной субтеломерной аномалией и оценили их клинические данные, семейный анамнез, анамнез родов, лицевые дисморфологические признаки и врожденные пороки [4]. Контрольную группу составили 110 детей с умственной отсталостью неясной этиологии, но без субмикроскопических субтеломерных перестроек. На основании этих исследований были разработаны показания для направления пациентов на исследование субмикроскопических субтеломерных перестроек. Такими показаниями являются семейный характер умственной отсталости; пренатальная гипотрофия; постнатальная задержка или опережение физического развития; наличие не менее двух малых аномалий и одной или более «нелицевой» малой аномалии и/или врожденного порока развития. В настоящий момент нет единого мнения относительно правильности и полноты этих рекомендаций. Для повышения эффективности диагностики субтеломерных нарушений необходимо выявление детальных клинических характеристик и применение методов исследования генома [4]. В последнее время появились новые методы, позволяющие измерить число копий локуса с помощью гибридизации со специфическим набором проб на концевых участках хромосом. С. Sismani и соавторы сообщили о так называемом MAPH-методе, использованном ими для скрининга с целью выявления субтеломерных перестроек. Этот метод является более быстрым и экономически выгодным, что позволяет рекомендовать его для скрининга субтеломерных перестроек. Отдельную проблему в медико-генетическом консультировании представляют пациенты с подозрением на Х-сцепленные формы генетических патологий, которые встречаются в популяции со средней частотой 0,15%. В последнее время на хромосоме Х было идентифицировано несколько генов, ответственных за неспецифическую умственную отсталость: мутации в гене TM4SF2 interleukin-1 семейства рецепторов IL1RAPL1 и в гене VCX-A; мутации гомеобоксного гена ARX и гена L1CAM при Х-сцепленной гидроцефалии. Недавно описан синдром МЕНМО включающий в себя умственную отсталость, эпилептические приступы, гипогенитализм, микроцефалию и ожирение. Ген, определяющий это состояние, локализован в районе Хр21.1-р22.13. Наиболее распространенной формой Х-сцепленной умственной отсталости является синдром Мартина-Белла, или синдром фрагильности Х-хромосомы (FRA-X). Известно, что частота синдрома Мартина-Белла в общей популяции мальчиков составляет 0,05-0,025%, а среди умственно отсталых пациентов мужского пола его частота значительно выше и колеблется, по разным данным, от 1,6 до 22%. Новые возможности для диагностики синдрома Мартина-Белла появились с развитием молекулярно-генетических методов исследования. Оказалось, что причиной возникновения синдрома является экспансия тринуклеотидного повтора CGG в генах FMR1 и FMR2. Много сообщений подтверждают общий клинический опыт о необходимости хромосомного анализа у пациентов с неспецифической, несиндромальной умственной отсталостью для выявления анеуплоидии по половым хромосомам и FRA-X. В исследовании А. Battaglia и соавт. у 10,2% детей с задержками развития была выявлена анеуплоидия, а у 5,1% была диагностирована FRA-X. М. Khalifa и соавт. обследовали 1205 пациентов с умственной отсталостью и выявили среди них 8 мальчиков с синдромом Клайнфельтера и 3 мальчиков с синдромом FRA-Х. В 1999 г. впервые была описана, а затем подтверждена другими исследованиями мутация в гене метил-CpG связывающего белка 2 (MECP2), вызывающая синдром Ретта [4].
Таким образом, для выявления генетических патологий, в настоящее время, стали широко использовать не только цитогенетические, но и молекулярно-цитогенетические методы исследований. Это позволяет более полно изучить проблемы наследственных заболеваний.
1.2 Геномный импринтинг
В середине XIX в. Грегор Мендель в своих опытах по скрещиванию гороха сделал наблюдение, которое впоследствии стало настоящей аксиомой для генетиков. Он обнаружил, что, если скрестить гомозиготное растение, имеющее гладкие семена, и гомозиготное растение с морщинистыми семенами, в потомстве все растения будут одинаковыми и дадут только гладкие семена. Этот результат не зависел от того, у какого из растении, взятых для скрещивания, - мужского или женского - семена были гладкими. Так Мендель открыл принцип эквивалентности реципрокных скрещиваний: у потомства ген действует одинаково независимо от того, от кого из родителей он унаследован.
Трудно переоценить значение этого наблюдения Менделя в истории и практике генетики. Было установлено, что такой закономерности подчиняется большое число наследственных признаков - не только у гороха, но и у многих других организмов [3].
Исключения из правила идентичности гибридов при реципрокных скрещиваниях (т. е. скрещиваниях между двумя формами, когда каждая из них в одном случае берется в качестве матери, а в другом - в качестве отца) в действительности известны давно, однако, как правило, их можно было отнести к одному из двух классов. Первый составляют признаки, которые определяются генами, расположенными в половых хромосомах: у самок млекопитающих в ядрах клеток имеется по две Х-хромосомы, у самцов - по одной Х- и одной У-хромосоме. Например, цветовая слепота и гемофилия связаны с генами X-хромосомы. Наследование этих сцепленных с полом признаков подчиняется вполне определенным правилам, согласно которым гибриды в реципрокных скрещиваниях не обязательно идентичны. Рассмотрим пример: у отца-дальтоника и нормальной по этому признаку матери ни один из сыновей не будет дальтоником. Если мать страдает дальтонизмом, а отец нет, то все сыновья окажутся дальтониками. В обоих случаях дочери будут нести ген, обусловливающий дальтонизм, но иметь нормальное зрение. Наследование и проявление признаков, сцепленных с полом, зависят от пола потомка, но не связаны непосредственно с полом того родителя, от которого унаследован признак.
Второй класс неэквивалентных реципрокных скрещиваний включает признаки, определяемые внеядерными генами. Некоторые клеточные органеллы - митохондрии в клетках животных, митохондрии и хлоропласты в клетках растений - обладают своей собственной генетической информацией. Эти органеллы передаются из поколения в поколение с цитоплазмой яйцеклеток и поэтому наследуются исключительно по материнской линии. Таков характер наследования цвета листьев у некоторых растений, а также заболевания человека, известного под названием митохондриальной энцефаломиопатии. При митохондриальном наследовании зигота, образующаяся в результате слияния половых клеток, получает митохондрии и содержащуюся в них мтДНК только через яйцеклетку [3].
Недавно генетики и эмбриологи описали третье исключение — это геномный импринтинг, когда оба родителя передают потомкам совершенно идентичные гены, но эти гены несут специфический отпечаток пола родителей, т.е. отцовские и материнские гены активированы или супрессированы во время гаметогенеза по-разному. Таким образом, в некоторых случаях важно, от кого из родителей унаследован ген [1]. Суть геномного импринтинга заключается в том, что гены, передаваемые потомству, несут специфический «отпечаток» пола родителя, т.е. отцовские и материнские гены маркированы по-разному; причем эти «отпечатки» временные и могут быть «стерты». Вследствие геномного импринтинга потомки, получившие маркированные гены от матери, отличаются от тех, которые унаследовали такие гены от отца. Другими словами, в некоторых случаях важно, от которого из родителей унаследован ген [1, 3].
Многие исследователи пытались установить молекулярную природу геномного импринтинга, обеспечивающие его механизмы, а также число и функции маркируемых генов. Благодаря этому сделано несколько замечательных открытий, которые расширяют понимание ряда раковых и наследственных заболеваний, а также некоторых других патологий. Изучение геномного импринтинга, возможно, откроет что-то новое и в наследовании признаков, которые вполне удовлетворительно объясняются в рамках классической менделевской генетики [3].
Термин «импринтинг» (imprint — отпечаток) впервые предложил в 1960 г. Х. Кроуз из Колумбийского университета США для описания селективной элиминации отцовских хромосом у насекомых. Геномный импринтинг называют эпигенетическим явлением, подчеркивая этим, что наследуются изменения генной активности, обусловленные родительским происхождением хромосом или их фрагментов, а не структурные перестройки генетического материала (мутации). Таким образом, в некоторых участках генома, подверженных геномному импринтингу, экспрессируется только один отцовский или материнский аллель, т.е. наблюдается моноаллельная экспрессия импринтированных генов (генов, которые дифференциально экспрессируются в зависимости от отцовского или материнского происхождения) в отличие от обычной диаллельной. Причем, если импринтирован материнский ген, то экспрессируется отцовский аллель и наоборот. Наличие такого способа регуляции работы генов свидетельствует о неэквивалентном вкладе родителей в функционирование генома потомков, а фенотипические признаки, контролируемые импринтированными локусами, могут появляться в результате не только мутаций генов, но и нарушения эпигенетической программы регуляции генной экспрессии [1].
Геномный импринтинг занимает особое место среди специфических механизмов регуляции активности генов на ранних стадиях развития, приводя к различиям в экспрессии гомологичных материнских и отцовских аллелей. Первоначальный «отпечаток», созданный в половых клетках, служит основанием для дальнейших модификаций в результате взаимодействий между родительскими геномами и цитоплазматическими факторами яйцеклетки во время формирования пронуклеуса (автономное существование яйцеклетки и сперматозоида в зиготе). Последующие эпигенетические модификации могут привести к тому, что изменения в экспрессии генов будут стабильно передаваться в процессе развития клеточных поколений. Геномный импринтинг, например, может изменять дозу генов, контролирующих рост эмбриона, клеточную пролиферацию и дифференцировку [1].
Изучение геномного импринтинга у млекопитающих началось в начале 80-х годов XX в. после опытов на мышах, проведенных Дж. Мак Гратом, Д. Солтером и М. Сурани (рис. 1). Авторы разработали тонкий микрохирургический метод переноса клеточных ядер мышиных эмбрионов в стадии пронуклеусов и показали, что наследование хромосомных наборов только от одного из родителей приводит к нарушению процесса развития. Оказалось, что отцовский генетический вклад важен для развития плаценты, а материнский вклад необходим для развития тела эмбриона. Далее было показано, что наследование части индивидуальных хромосом или целой хромосомы только от одного из родителей может также приводить к аномальному фенотипу [3].
Таким образом, геномный импринтинг состоит в том, что хромосомы половых клеток (сперматозоидов или яйцеклеток) индивида приобретают «отпечаток» его пола (рис. 2). Потомство получает один набор хромосом с отцовской маркировкой некоторых генов, а другой - с материнской. При образовании у потомка половых клеток прежний «отпечаток» стирается и эти гены маркируются в соответствии с полом данной особи.
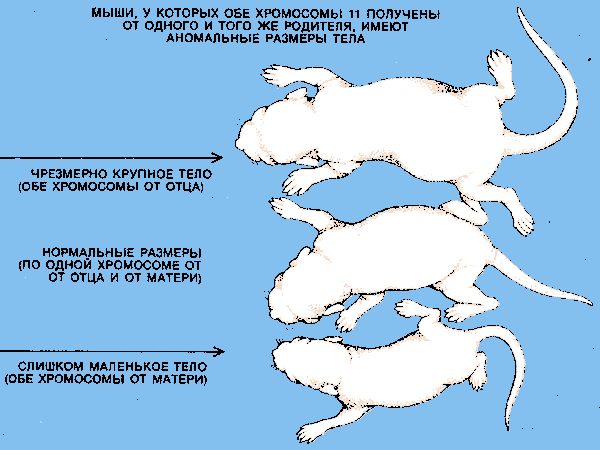
Рис. 1 Геномный импринтинг у мышей
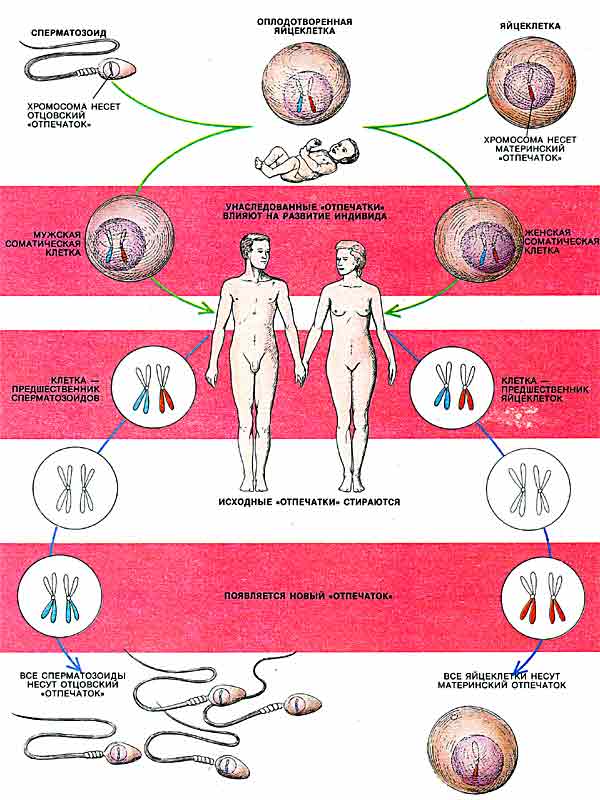
Рис. 2 Геномный импринтинг у человека
1.3 Болезни импринтинга
Проявления геномного импринтинга удивительным образом связаны с некоторыми заболеваниями человека. Неожиданно оказалось, что в природе уже существуют параллели тем экспериментальным состояниям, которые исследовали у мышей. Недавно Р. Николлс и его коллеги из Медицинской школы Гарвардского университета установили, что у многих больных с синдромов Прадера—Вилли обе хромосомы 15 унаследованы от матери [3].
Р. Николлс, Дж. Кнолл (тоже сотрудник Гарвардского университета) и Ч. Уильямс из Флоридского университета обнаружили связанную с геномным импринтингом закономерность у больных с синдромом Ангельмана. У таких больных нередко имеют место частичные делеции унаследованной от матери хромосомы 15, в результате чего полностью функциональна только отцовская хромосома 15.
Эти два заболевания, хотя и столь разные по клинической картине, могут быть связаны с различиями в импринтинге одних и тех же генов одной и той же хромосомы. Однако в отличие от ненормально крупных и мелких мышей, синдромы Прадера—Вилли и Ангельмана не удается представить просто как две стороны одной медали, т. е. объяснить избытком либо недостатком продуктов одних и тех же генов. Исследования Николлса и его коллег показали, что бывает очень трудно предсказать, каким образом конкретные признаки зависят от процесса геномного импринтинга. Полезно было бы изучить на этот предмет многие наследственные заболевания человека; не исключено, что найдутся указания на влияние геномного импринтинга [3].
Примером импринтинга целого генома у человека является истинный пузырный занос, который возникает при оплодотворении яйцеклетки, лишенной материнских хромосом, двумя сперматозоидами. Несмотря на наличие полноценного диплоидного набора, ранний эмбриогенез таких зигот протекает аномально: ткани собственно эмбриона вообще не формируются, однако бурно разрастается трофобласт. В случае двойного набора материнских хромосом развивается тератома — эмбриональная опухоль. Следовательно, у человека, как и у мыши, на ранних стадиях развития геном отца преимущественно обеспечивает развитие провизорных органов, а геном матери — эмбриональных структур. Только материнский или только отцовский геномы не в состоянии обеспечить нормальное развитие эмбриона [1, 3].
На организменном уровне эффект импринтинга обнаружен в связи с наличием в хромосомном наборе фрагментов или целых хромосом одного (материнского или отцовского) происхождения — так называемая однородытельская дисомия (ОРД), т.е. наблюдается качественный, а не количественный хромосомный дисбаланс. Известны два основных механизма образования ОРД у человека: коррекция трисомии до дисомии (гетеродисомия), происходящая в 1-м мейотическом делении, и коррекция моносомии до дисомии (изодисомия) — во 2-м мейотическом делении.
Феноменология импринтинга значительно лучше изучена у мыши, чем у человека, и поскольку известна гомология между хромосомами человека и мыши (примерно по 700 локусам), можно использовать данные по импринтингу, полученные на мышах, для целенаправленного поиска импринтинга по определенным локусам у человека.
Импринтированные гены и их транскрипты обнаружены на многих хромосомах человека — 1, 5, 6, 7, 11, 13, 15, 19, 20 и X. На хромосоме 7 мыши и хромосомах 1 и 15 человека найдены два больших кластера ортологичных импринтированных генов, т.е. эволюционно консервативных по статусу импринтинга. Идентифицированы гены с полиморфным импринтингом, т.е. с сочетанием моноаллельной экспрессии в одних тканях и диаллельной — в других. По-видимому, такая тканеспецифическая эпигенетическая модификация некоторых генов может быть одним из механизмов, обеспечивающих дифференциальную экспрессию генов клеток разных тканей в ходе развития [1].
В последние годы с помощью молекулярно-генетических методов феномен геномного импринтинга наблюдают и при мультифакториальных заболеваниях. Например, четко выраженный отцовский импринтинг обнаружен при атопическом дерматите, материнский — при бронхиальной астме и атопии у детей. При инсулинзависимом сахарном диабете выявлена более высокая вероятность отцовского импринтинга. Ген инсулина у человека расположен в кластере импринтированных генов 11р15 и гомологичен локусам в мышином геноме, подверженным импринтингу. Кроме того, обнаружена ОРД отцовского происхождения у детей с неонатальным сахарным диабетом.
Примеров заболеваний, в основе этиологии которых лежит нарушение функции импринтированных участков генома, довольно много, поэтому можно говорить об особом классе заболеваний человека — «болезнях импринтинга», которых насчитывается уже более 30. Основные из них приведены в табл. 1.
Таблица 1
Предполагаемые «болезни импринтинга» у человека
|
Заболевание |
Хромосома |
Происхождение |
|
Синдром Адамса—Оливера. |
Материнское |
|
|
Болезнь Альцгеймера |
Отцовское |
|
|
Синдром Энжельмена |
15 |
Материнское |
|
Атопия |
11 |
То же |
|
Церебеллярная атаксия |
Отцовское |
|
|
Расщелина губы |
То же |
|
|
Врожденный порок сердца |
Материнское |
|
|
Семейные опухоли клубочков |
11 |
Отцовское |
|
Синдром ломкой хромосомы X |
X |
Материнское |
|
Синдром Гольденхара |
То же |
|
|
Хорея Гентингтона (ювенильная форма) |
4 |
Отцовское |
|
Идиопатический гипертрофический субаортальный стеноз |
То же |
|
|
Злокачественная гипертермия |
19 |
Материнское |
|
Миотоническая дистрофия (врожденная) |
19 |
То же |
|
Нарколепсия |
6 |
» » |
|
Дефекты невральной трубки |
Отцовское |
|
|
Нейрофиброматоз 1 |
17 |
Материнское |
|
Нейрофиброматоз II |
22 |
То же |
|
Поликистоз почек (два локуса) |
16 и ? |
Материнское и отцовское |
|
Поликистоз яичников |
Материнское |
|
|
Синдром Прадера—Вилли |
15 |
Отцовское |
|
Псориаз |
То же |
|
|
Псевдопсевдогипопаратиреоз |
20 |
Материнское |
|
Спиноцеребеллярная атаксия |
Отцовское |
|
|
Туберозный склероз |
Материнское |
|
|
Синдром Видемана—Беквита |
11 |
То же |
|
Билатеральная спорадическая ретинобластома |
13 |
» » |
|
Агенезия почек, аномалии лица |
16 |
» » |
|
Синдром лицевых аномалий, микрокрании, аномалий респираторного тракта, гепатомегалии |
14 |
Отцовское |
|
Синдром Сильвера—Рассела |
7 |
Материнское |
|
Синдром умственной отсталости, низкого роста, преждевременного полового созревания |
14 |
То же |
|
Заболевание |
Хромосома |
Происхождение |
|
Синдром Адамса—Оливера. |
Материнское |
|
|
Болезнь Альцгеймера |
Отцовское |
|
|
Синдром Энжельмена |
15 |
Материнское |
|
Атопия |
11 |
То же |
|
Церебеллярная атаксия |
Отцовское |
|
|
Расщелина губы |
То же |
|
|
Врожденный порок сердца |
Материнское |
микроделеция дисомия наследственный импринтинг
ГЛАВА 2. ХАРАКТЕРИСТИКА СИНДРОМОВ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА
2.1 Цитогенетическая характеристика синдромов Прадера-Вилли и Ангельмана
Наиболее убедительные данные получены при синдроме Прадера—Вилли (СПВ) и синдроме Ангельмана (СЭ), которые, имея существенно разные клинические проявления, в своей основе имеют сходные молекулярно-цитогенетические изменения (табл. 2 и 3).
Таблица 2
Корреляция генотип-фенотип при синдроме Ангельмана
|
Симптом |
Del |
ОРД (2-3 %) |
Мутации |
|
15q11-q13 (70-75 %) |
«импринтинга» (3-4 %) |
||
|
Тяжелая умственная отсталость |
+ |
+ |
+ |
|
Отсутствие речи |
+ |
+ |
+ |
|
Атаксия |
+ |
+ |
+ |
|
Судороги |
+ |
+ |
+ |
|
Пароксизмы смеха |
+ |
+ |
+ |
|
Гиперактивность |
+ |
+ |
+ |
|
Аномалии ЭЭГ |
+ |
+ |
+ |
|
Характерное лицо |
+ |
+ |
+ |
|
Гипопигментация |
+ |
+ |
— |
Таблица 3
Корреляция генотип—фенотип при синдроме Прадера-Вили
|
Симптом |
Del |
ОРД (20-25 %) |
Мутации |
|
15q11-q13 (70-75 %) |
«импринтинга» (3-4 %) |
||
|
Мышечная гипотония |
+ |
+ |
+ |
|
Ожирение |
+ |
+ |
+ |
|
Полифагия |
+ |
+ |
+ |
|
Характерное лицо |
+ |
+ |
+ |
|
Умственная отсталость |
+ |
+ |
+ |
|
Гипогонадизм |
+ |
+ |
+ |
|
Акромикрия |
+ |
+ |
+ |
|
Низкий вес при рождении |
— |
+ |
— |
|
Низкий рост при рождении |
— |
+ |
— |
|
Гипопигментация |
+ |
+ |
— |
Наиболее частой причиной возникновения СПВ и СА является протяженная (до 4 млн. п.н.) деления критического района 15(q11—q13), которую находят у 70—75 % больных с этими синдромами. Делецию при СПВ обнаруживают на отцовской хромосоме 15, а при СЭ делеция той же области на ее материнском гомологе. Второй причиной возникновения СПВ и СЭ оказалась однородительская дисомия, т.е. наследование обоих гомологов хромосомы 15 от одного из родителей. С помощью ДНК-маркеров региона делеции путем блотт-гибридизации по Саузерну, а также анализа метилирования было продемонстрировано различное родительское происхождение хромосомы 15: в первом случае отцовское, во втором — материнское. Поскольку этот регион хромосомы 15 идентичен аналогичному региону хромосомы 2 мыши, для которого хорошо известен геномный импринтинг, исследования стали проводить в этом направлении. Оказалось, что регион СПВ активен на отцовской хромосоме (при наличии делеции на отцовской хромосоме или материнской ОРД отсутствуют отцовские гены) и не активен на материнской (рис. 3). Из рис. 1 видно, что при СПВ не экспрессируются отцовские гены, а при СЭ — материнские гены. Материнская ОРД наблюдается в 25 % случаев СПВ, а отцовская ОРД становится причиной возникновения СЭ в 3—5 % случаев [1].
В последние годы появились сообщения еще об одной причине развития этих синдромов у пациентов, у которых не было найдено ни типичных делеций, ни ОРД, но зато в семьях таких больных встречались повторные случаи синдрома.
В ходе исследования в проксимальном участке хромосомы 15 были обнаружены противоположно импринтированные гены — кандидаты СПВ и СЭ, соответственно SNRPN и UBE3A, в которых выявили мутации.
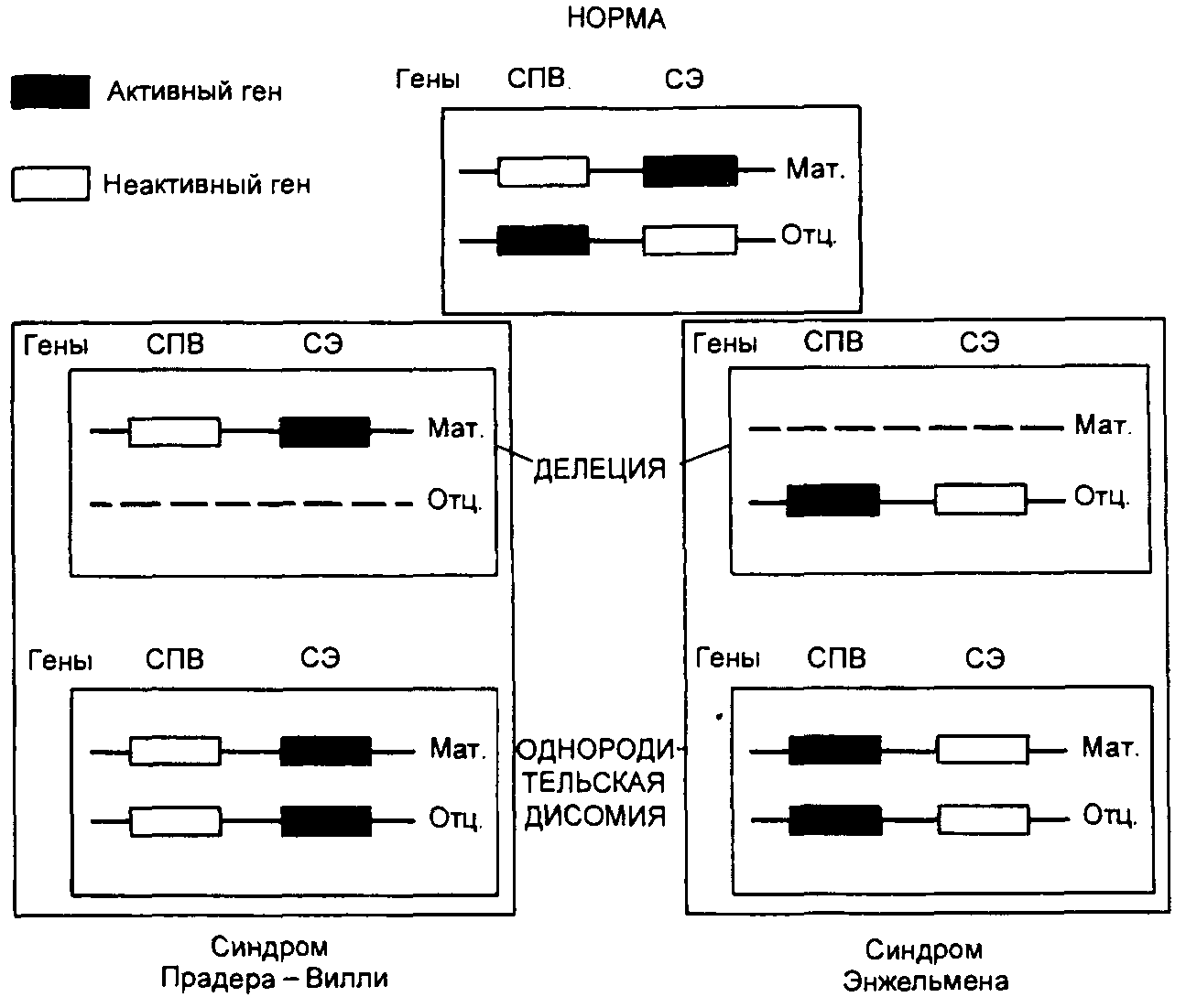
Рис. 3 Механизмы возникновения синдромов Прадера—Вилли и Ангельмана через делецию участка хромосомы 15 или однородительскую дисомию по хромосоме 15. СПВ — синдром Прадера—Вилли; СА — синдром Ангельмана; мат. — материнская хромосома; отц. — отцовская хромосома
Ген SNRPN кодирует полипептид N малого ядерного рибонуклеопротеина, активно экспрессируется исключительно на отцовской хромосоме 15 и репрессирован на материнском гомологе, т.е. мутации в этом гене вовлечены в патогенез СПВ. Критический регион для СЭ расположен дистальнее (локус D15S10), который экспрессируется только в материнских хромосомах. Предполагают, что мутации при СЭ есть в гене UBE3A кодирующем убиквитинлигазный белок ЗА. Экспрессия этого белка выявлена во всех тканях человека, причем в ряде структур мозга ген UВЕЗА активен лишь на материнской хромосоме. Дефицит материнской копии этого гена в клетках Пуркинье (грушевидных невроцитах мозжечка) и нейронах гиппокампа может, по-видимому, объяснить клиническую картину СЭ (умственная отсталость, атаксия, тремор и др.). Таким образом, в районе хромосомы 15(q11—q13) имеются близко расположенные, но противоположно импринтированные локусы, отвечающие за возникновение этих двух синдромов.
Эта область хромосомы 15 чрезвычайно существенна для нормальной переустановки геномного импринтинга. Она названа центром импринтинга (IC). Мутации в данной области приводят к ошибкам импринтинга, т.е. теряется способность стирать отпечаток предшествующего поколения. Так, если в сперматогенезе отца не происходит замены «женского» импринта на «мужской» на его материнской хромосоме, то в следующем поколении возникнет состояние, аналогичное материнской ОРД, которое будет сопровождаться фенотипом СПВ. Нарушение установления «женского» эпигенотипа на отцовских хромосомах в овогенезе матери приведет к развитию СА у потомства.
Повторный риск для трех групп семей при СПВ и СЭ будет существенно различаться. Так, при делециях он будет ниже 1 %, при ОРД риск также низкий, но в этом случае нужно учитывать возраст матери, который может увеличивать риск. При мутациях в центре импринтинга повторный риск будет существенно выше не только для родителей больного, но и ближайших родственников.
Связь геномного импринтинга с другой наследственной патологией человека на уровне хромосом или отдельных генов также отчетливо прослеживается и в настоящее время широко изучается. Так, например, при хорее Гентингтона и спинно-мозжечковой атаксии I заболевание возникает раньше и протекает тяжелее, если унаследованные гены имеют отцовское происхождение. При нейрофиброматозе 1 и 2, миотонической дистрофии, наоборот, заболевание имеет более раннее начало и тяжелое течение при унаследовании мутантных генов от матери. Не вызывает сомнения причастность геномного импринтинга к этиологии опухолевого роста. Выключение импринтинга, а также потеря гетерозиготности или ОРД по хромосомам или их участкам, содержащим импринтированные локусы, могут приводить к функциональной нуллисомии генов-супрессоров опухолевого роста или к аберрантной экспрессии протоонкогенов, что может лежать в основе возникновения рака. Кроме того, вероятность ОРД повышается не только с возрастом матери, но и у носителей изохромосом, робертсоновских и реципрокных транслокаций. Следует иметь в виду, что ОРД (изодисомия) может привести к гомозиготизации определенных регионов хромосомы и быть причиной аутосомно-рецессивной патологии. Такие случаи описаны, например, при муковисцидозе.
Точные механизмы, лежащие в основе дифференциальной экспрессии материнских и отцовских геномов, пока не известны. Основную роль в этом процессе отводят специфическому метилированию цитозиновых оснований ДНК. Важнейшими особенностями метилирования ДНК являются, во-первых, стабильное сохранение в ряду многих поколений клеток, а во-вторых, прямое или косвенное влияние на экспрессию генов. Специфическое для пола метилирование некоторых участков генома устанавливается во время гаметогенеза. Известно, что некоторые повторяющиеся и даже уникальные последовательности ДНК являются недометилированными в яйцеклетках и гиперметилированными в сперматозоидах. Такие различия между родительскими хромосомами сохраняются и после оплодотворения и стабильно передаются в следующие клеточные поколения. Как правило, активный ген ассоциируется со сниженным метилированием или его отсутствием, а неэкспрессирующий генетический регион — с гиперметилированием. Тканеспецифичное метилирование цитозиновых остатков ДНК осуществляется в ходе гамето- и эмбриогенеза с помощью ДНК-метилтрансфераз.
Значительная доля импринтированных генов (до 15 %) ассоциирует с антисмысловыми транскриптами. Такие транскрипты представлены обычно антисмысловой РНК, происходящей из интронов некоторых генов, и колинеарной ДНК. Эта антисмысловая РНК не выполняет кодирующих функций и, возможно, является регуляторной. Предполагают, что существуют и другие механизмы, регулирующие дифференциальную активность отцовских и материнских генов.
Описывают две модели смены эпигенотипа хромосом в гаметогенезе. Согласно первой, переключение эпигенотипа происходит только в той из гомологичных хромосом, которая унаследована от родителя противоположного пола, а вторую хромосому модификации не затрагивают. Вторая модель предполагает предварительное устранение («стирание») существующего эпигенотипа на обеих родительских хромосомах с последующим установлением импринта, соответствующего данному полу. За последние годы в результате многочисленных исследований метилирования и функционирования импринтированных генов в клетках зародышевого пути были получены убедительные доказательства в пользу второго предположения.
Таким образом, хотя роль метилирования в обеспечении аллельспецифической экспрессии генов несомненна, остается неясным, является ли метилирование первичным эпигенетическим сигналом, который «стирается» и устанавливается в га-метогенезе, или представляет собой некий вторичный процесс по отношению к более ранней стадии импринтинга и служит лишь для поддержания ранее установленного импринта.
Хотя в настоящее время не вызывает сомнения, что метилирование ДНК является эпигенетической меткой и оно достаточно хорошо изучено и характерно практически для всех импринтированных генов и локусов, нельзя исключить и другие пока еще неизвестные механизмы. Дальнейшее изучение геномного импринтинга (особенно в рамках функциональной геномики) будет иметь существенное значение для понимания тонких механизмов регуляции генной активности в онтогенезе и его связи с патологией человека [3].
Импринтинг генов ведет к необычным последствиям. У мужчин материнская копия хромосомы 15 содержит в себе знак того, что она пришла от матери. Но уже в следующем поколении у дочери или сына эта же хромосома будет содержать знак отцовского происхождения. В какой-то момент должно произойти переключение знака хромосомы на противоположный. Нет сомнений в том, что такое переключение происходит, поскольку только этим можно объяснить синдром Ангельмана. Никаких видимых повреждений на хромосоме 15 нет, просто две хромосомы ведут себя так, как будто обе произошли от отца. Это объясняется тем, что в нужный момент в организме матери не произошло переключение знака хромосомы. Возникновение данной проблемы можно проследить в поколениях и обнаружить мутацию в небольшом участке ДНК, непосредственно примыкающем к диверсифицированным генам. Это так называемый центр импринтинга, который каким-то образом указывает на происхождение хромосомы. Импринтинг генов осуществляется с помощью метилирования.
2.2 Клинические проявления и методы диагностики синдромов Прадера-Вилли и Ангельмана
2.2.1 Клинические проявления синдрома Прадера-Вилли
Синдром Прадера-Вилли (синдром Прадера-Лабхарта-Вилли-Фанкони, синдром НННО) описан в 1956 году является наиболее частой причиной генетически обусловленного тяжело протекающего ожирения у детей старше 1 года жизни и у взрослых. Встречается с одинаковой частотой среди обоих полов, регистрируется у лиц разных национальностей и рас. Популяционная частота составляет от 1:10 000 до 1:15 000 [5, 6].
Клиническая диагностика СПВ на 1-м году жизни затруднительна в силу отсутствия специфической симптоматики при рождении и быстрой спонтанной ликвидации ранних признаков заболевания после первых месяцев жизни.
К этим ранним проявлениям синдрома относятся:
- вялое шевеление плода,
- слабость сосательного рефлекса вскоре после рождения,
- развитие мышечной гипотонии в первые месяцы жизни.
Уже на 1-м году жизни могут выявляться различные дизморфии лица и конечностей, гипогонадизм и/или гипогенитализм. Наличие у ребенка первых месяцев жизни указанных симптомов нередко приводит врача к ошибочной диагностике пре- и перинатального поражения ЦНС, инфекционного процесса, так как часты нарушения терморегуляции; различных вариантов наследственных миопатий, врожденного гипотиреоза и других болезней. В существенной степени диагностику СПВ на 1-м году жизни затрудняют спонтанное восстановление сосательного рефлекса в первые месяцы жизни и повышение мышечного тонуса. Вместе с тем, развитие дефицитных заболеваний и необоснованное лечение по поводу другой предполагаемой патологии могут сказаться на последующем онтогенезе ребенка, в частности, его интеллектуальном развитии. Этот факт, несомненно, определяет актуальность ранней диагностики СПВ [5].
В большинстве случаев клиническая диагностика СПВ осуществляется после годовалого возраста ребенка, когда развивается 2-я фаза заболевания, характеризующаяся появлением у больного постоянного чувства голода. У ребенка постепенно и стойко формируется поведение «постоянного поиска пищи». Быстро развивается и прогрессирует ожирение, замедляются процессы роста, появляются умеренные признаки нарушения интеллекта, в дальнейшем нарушаются сроки полового развития и порядок появления вторичных половых признаков. Часто присутствует дневная сонливость, нередко по типу нарколепсии, а также ночные апноэ. В этой связи больные СПВ на протяжении своей жизни подвержены синдрому внезапной смерти. В патогенезе развития 2-й фазы заболевания основную роль играет гипоталамическая дисфункция, в том числе дефицит СТГ (гормона роста). Средняя окончательная длина тела без лечения СТГ у мальчиков составляет 155 см, у девочек — 147 см. Диагностика СПВ до развития 2-й фазы заболевания весьма актуальна. Она создает предпосылки для своевременного формирования у ребенка правильного поведения, в частности, пищевого. Коррекция дефицита СТГ, начатая до 18-месячного возраста ребенка, способствует формированию у него правильного телосложения, препятствует избыточному жироотложению, существенно улучшает развитие мышечной моторики. Последнее время в литературе ведется полемика о целесообразности длительной терапии соматотропином при СПВ.
Возможности ранней диагностики СПВ в существенной степени повышаются при использовании диагностических методов, в основе которых лежит балльная оценка присутствующих у больного больших и малых признаков.
К большим признакам (каждому присваивается 1 балл) относятся следующие:
• характерные лицевые симптомы (долихоцефалия с уменьшением битемпорального диаметра, миндалевидный разрез глаз, небольшой рот с опущенными вниз углами и тонкой верхней губой, страбизм);
• задержка нервно-психического развития до 6 лет, умеренное снижение интеллекта и проблемы обучения в школьном возрасте;
• проблемы при кормлении в первые месяцы жизни с последующей нормализацией сосания в течение грудного периода;
• изменения со стороны половой сферы;
• мышечная гипотония центрального генеза в раннем детстве;
• прогрессирующее ожирение в возрасте от 1 года до 6 лет.
К малым признакам (каждому присваивается 0,5 балла) относятся следующие:
• снижение двигательной активности плода и инфантильная летаргия;
• нарушения рефракции;
• снижение пигментации кожи и волос в сравнении с родителями;
• следы «потертостей» кожных покровов;
• поперечная ладонная складка;
• низкорослость к 15 годам с учетом длины тела других членов семьи;
• нарушения сна и апноэ во время сна;
• маленькие стопы и/или кисти;
• дефекты артикуляции и речи;
• густая, вязкая слюна;
• поведенческие нарушения [2, 5, 6].
Клинический симптомокомплекс Синдрома Прадера-Вилли включает сахарный диабет или нарушение толерантности к глюкозе. Наличие таких важных диагностических симптомов, как мышечная гипотония (Hypotonia), умственная отсталость (Hypomentia), гипогонадизм (Hypogonadism) и ожирение (Obesity), послужило основанием для одного из наименований синдрома – НННО.
Тяжелая мышечная гипотония является наиболее ранним симптомом заболевания, возникает уже во внутриутробный период, что объясняет снижение подвижности плода. В ранний постнатальный период имеют место снижение сухожильных, глотательного и сосательного рефлексов, затрудняющих кормление, дыхательные нарушения, малоподвижность, задержка развития двигательных функций.
Со второго полугодия жизни мышечная гипотония заметно уменьшается, однако и у взрослых может сохраняться снижение мышечного тонуса. Появляется полифагия, развивается ожирение. Характерно отложение жира преимущественно в области туловища и проксимальных отделов конечностей, на этом фоне кисти и стопы кажутся диспропорционально маленькими (акромикрия). Акромикрия сочетается с клинодактилией, синдактилией, брахимезофалангией.
Гипогонадотропный гипогонадизм у лиц мужского пола приобретает клиническую выраженность к пубертатному периоду и сохраняется у взрослых. Его особенности – резкое недоразвитие гениталий, скудное вторичное оволосение, снижение либидо и потенции, атрофия тестикулярной ткани, снижение сперматогенеза. Уже с рождения у мальчиков выявляют двусторонний крипторхизм, маленькую, гладкую мошонку и резкую гипоплазию полового члена, часто с фимозом.
У лиц женского пола обнаруживают гипоплазию половых губ, позднее появление вторичных половых признаков, задержки менструаций вплоть до аменореи, инфантилизм матки, бесплодие. Больные обоих полов обычно стерильны.
Психомоторное развитие детей замедлено, у большинства больных имеется различной формы умственная отсталость, в редких случаях отмечен нормальный или субнормальный интеллект. Больные, как правило, доброжелательны, безинициативны, плохо контролируют свои эмоции, им свойственна резкая смена настроения [6].
К специфическим черепно-лицевым дизморфиям относятся нерезко выраженная микроцефалия, гипоплазия хрящей ушных раковин, деформация и низкое расположение ушей, сдавленный в височных областях лоб, высокое арковидное небо, гипоплазия нижней челюсти, микродонтия с дефектами эмали и кариесом.
Примерно у половины больных наблюдаются гипопигментация кожи, волос и радужки, некоторое повышение фоточувствительности.
У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы - коэффициент интеллектуального развития - от 20 до 80 ед. (при норме 85-115 ед.). Речь затруднена, словарный запас уменьшен.
Встречаются и другие аномалии: микродонтия, сколиоз, эктропион (выворот века), глаукома.
При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга.
Продолжительность жизни больных может достигать 60 лет и более.
Согласно данным литературы, патогенез синдрома Прадера-Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза.
Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков [5].
Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке.
Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера-Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может оказать роковое влияние на развитие злокачественных новообразований у лиц с синдромом Прадера-Вилли [5].
Терапия синдрома Прадера-Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины).
СПВ должен быть заподозрен у детей до 3-летнего возраста при наличии не менее 5 баллов, а у детей старше 3 лет — 8 баллов, при условии присутствия 4 и более больших признаков.
Несмотря на высокую диагностическую чувствительность приведенной шкалы (около 90%), во всех случаях для подтверждения диагноза требуются кариотипирование и молекулярно-генетические исследования 15-й пары хромосом. Специфичность этих исследований достигает 100% . Кроме того, от выявленного типа генетических нарушений зависит генетический прогноз потомства.
Дети, страдающие СПВ, должны постоянно находиться под наблюдением педиатра, невролога, психотерапевта, эндокринолога и офтальмолога.
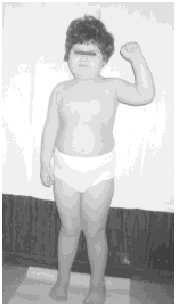
Рис. 4. Девочка с синдромом Прадера-Вилли
2.2.2 Клинические проявления синдрома Ангельмана
В 1965 году доктор Гарри Ангельман, английский психиатр, впервые описал трех детей с характеристиками, в настоящее время известными как синдром Ангельмана. Он отметил, что все эти дети имели неуклюжесть, трясущуюся походку, отсутствие речи, излишне смешливы и впадали в припадки. Другие похожие случаи уже были описаны учеными, но эти случаи были особенными, и многие психиатры подтвердили их исключительность. Первые отчеты пришли из Северной Америки в начале 1980-х гг.
Синдром Ангельмана — генетическая аномалия. Для него характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки.
Частота встречаемости, по разным данным, — 1: 10 000-20 000 живорожденных младенцев. (Однако, согласно данным Центра развития человека и отклонений в развитии (университет Вашингтона, США), можно предполагать, что доля людей с синдромом Ангельмана в действительности намного больше статистической.)
Синдром Ангельмана обычно не распознается при рождении или в раннем детстве, пока не проявят себя проблемы в развитии, которые не специфичны к этому времени. Родители могут заподозрить диагноз после прочтения о синдроме Ангельмана или после встречи ребенка с такими же признаками. Наиболее распространенный возраст для диагностики – между тремя и семью годами, когда отличительные черты поведения становятся более очевидными.
Для синдрома Ангельмана характерны:
- В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес;
- задержка в развитии навыков общей моторики (умение сидеть, ходить);
- задержка речевого развития, неразвитая речь (у всех детей);
- дети больше понимают, чем могут сказать или выразить;
- дефицит внимания и гиперактивность;
- сложности с обучением;
- эпилепсия (80% случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана происходит вторичная (симптоматическая) общая эпилепсия;
- необычные движения (мелкий тремор, хаотические движения конечностей);
- частый смех без повода;
- ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками;
- размер головы меньше среднего, нередко с уплощением затылка;
- иногда особые черты лица — широкий рот, зубы с промежутками между ними, выдающийся вперед подбородок, высунутый наружу язык);
- нарушения сна;
- страбизм (косоглазие) в 40 % случаев;
- сколиоз (искривление позвоночника) в 10 % случаев;
- повышенная чувствительность к высокой температуре;
- бывают сильно увлечены играми с водой [7].
Диагностика: синдром диагностируется путем генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи.
Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15q11-q13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE3A.
Существует небольшая группа людей, у которых результаты всех вышеописанных анализов в норме, однако наблюдаются все внешние проявления синдрома Ангельмана. Наука пока не дает ответ на вопрос, как это возможно.
Лечение: синдром Ангельмана является врожденной генетической аномалией и, следовательно, не может быть излечен.
Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом.
В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии).
Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом.
Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0.3 мг мелатонина за 30 минут-1 час перед сном улучшает сон пациентов с синдромом Ангельмана. А нарушения стула регулируются назначением легких слабительных. Приступы лечатся так же, как эпилепсия.
Дети с синдромом Ангельмана часто испытывают больше одного типа приступов. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложность в общении с другими людьми. Врачи США показывают, что для аутичных детей внутривенные инъекции гормона секретина успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана [2, 7].
Риски: оценка риска повторного рождения ребенка с синдромом Ангельмана у тех же родителей очень сложна, необходима консультация врача-генетика. Считается, что обычная делеция является спонтанной, риск повтора меньше 1 %. В случае молекулярной микроделеции в 15q11-q13, если она наблюдается и у матери, риск теоретически до 50 %. Мутации внутри гена UBE3A могут быть случайными и неунаследованными, в этой ситуации риск повтора <1%; однако эти мутации можно унаследовать от нормальной матери, и тогда теоретический риск 50%. Партеногенетическая дисомия 15-ой пары – случайная ситуация; риск повтора <1%. Есть несколько людей с AS с необычными преобразованиями хромосомы 15, включая область 15q11-q13; в этих случаях оценка риска повтора зависит от хромосомных нарушений у родителей [7].
Перспективы развития: дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарем 5-10 слов. При этом дети/люди с синдромом Ангельмана любят общаться с людьми, играть.
Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам, направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты.
Перспективы развития зависят от степени пораженности хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы).
С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки.
Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые — и ночью.
Некоторые люди с синдромом Ангельмана способны есть ножом и вилкой, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний».
Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом.
Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом [6, 7, 8].
Таким образом, несмотря на повреждение при синдромах Прадера - Вилли и Ангельмана одного и того же локуса хромосомы 15, клинические проявления обеих болезней резко противоположны.
2.3 Связь синдромов Прадера-Вилли и Ангельмана
Установлено, что формирование синдрома Прадера-Вилли и синдрома Ангельмана связано с геномным импринтингом [4]. В большинстве случаев синдрома Прадера-Вилли (70%) микроделеции района 15q11-q13, обнаруживаемые у пациентов, имеют отцовское происхождение. Примерно в 30% случаев у пациентов обнаруживаются только материнские аллели (однородительская материнская дисомия). Эта своеобразная «гомозиготизация» происходит в результате неравного кроссинговера или соматической рекомбинации по механизму конверсии генов. Синдром Ангельмана является как бы зеркальным отражением синдрома Прадера-Вилли. Делеции, захватывающие примерно тот же район хромосомы 15, что и при синдроме Прадера-Вилли, имеют материнское происхождение, а однородительская дисомия - отцовское. Установлено, что в перицентромерном участке хромосомы 15 имеется тандемный инвертированный повтор MN7 [4], который, по-видимому, определяет высокую мутабельность района 15q11.2. Пациенты с интерстициальной дупликацией проксимального района 15q могут иметь задержку развития без признаков дисморфогенеза. С целью определения степени выраженности патологии пациенты с дупликацией родительского происхождения критического региона 15q были разделены на 3 группы. К первой группе были отнесены больные с эухроматиновым вариантом дупликации без захвата локуса 15q11-q13. Вторая группа включала пациентов с дупликацией 15q11-q13 материнского происхождения, у которых имели место трудности обучения и речи, признаки аутизма или атипичный аутизм без дисморфологических черт. Третья группа включала пациентов с дупликациями отцовского происхождения без очевидного клинического фенотипа [4]. У одного больного, имевшего дупликацию 15q отцовского происхождения с вовлечением локуса 15q11-q13, были выявлены задержка развития речи и аномалия мозга. Инвертированная дупликация inv dup 15 с вовлечением локуса 15q11-q13 была обнаружена у детей с задержкой развития, умственной отсталостью от умеренной до тяжелой, эпилепсией, манифестировавшей в возрасте от 4 до 8 лет, генерализованной гипотонией и аутистическим поведением, но без дисморфологических черт или с 1-4 малыми аномалиями развития [4]. К настоящему времени описано два цитогенетических типа инвертированной дупликации хромосомы 15. Первый тип включает метацентрик или субметацентрик и гетерохроматиновую хромосому, меньшую или равную хромосомам G группы, - dic(15)(q11). Большинство детей с данной аномалией имеют нормальный фенотип. Второй тип включает хромосому большего размера, чем хромосомы группы G, и имеет 15q эухроматиновый участок. Данный участок включает 15q11-q13 регион и его цитогенетическое описание - dic(15)(q12 или q13). Этот дицентрик получен из двух соответственных материнских хромосом в мейозе и обычно связан с большим возрастом матери и аномальным фенотипом, описанным выше. Таким образом, при подозрении на данную хромосомную аномалию необходимо сочетать стандартный цитогенетический анализ с FISH-анализом. Задержка развития легкой степени с Прадер-Вилли-подобным фенотипом описана у пациентов с однородительской дисомией хромосомы 14 материнского происхождения. Однородительская дисомия хромосомы 14 отцовского происхождения также может быть ассоциирована с различными аномалиями. Однородительская дисомия хромосомы 14 является, таким образом, одной из возможных причин задержки развития и должна выявляться в подозрительных случаях методом ДНК-анализа. Это один из путей улучшения диагностики умственной отсталости. С. Williams и соавторы привлекли внимание к состояниям, по своим фенотипическим проявлениям напоминающим синдром Ангельмана. Данные наследственные формы могут быть вызваны как хромосомными аберрациями, так и генными мутациями. Могут отмечаться терминальные делеции 22q13.3, реже микродупликации 15q11-13, интерстициальные делеции 2q21-23, 17q23.3, 4q. По мнению автора, все эти хромосомные аномалии должны быть проанализированы при обследовании пациентов с фенотипическими особенностями, напоминающими синдром Ангельмана. Одним из синдромов, требующих дифференциальной диагностики с синдромом Ангельмана, является синдром Ретта, особенно учитывая, что многие пациенты с данным состоянием умирают в младенчестве или раннем детстве. Отсутствием речи, атаксией, эпилептическими приступами и поведенческими особенностями, характерными для синдрома Ангельмана, может проявляться дефицит метилен-тетрагидрофолат-редуктазы. Глубокая умственная отсталость и протрузия языка могут быть проявлениями АТR-Х синдрома (Х-сцепленный рецессивный синдром альфа-талассемии).А. Battaglia и соавторы приводят алгоритм полного генетического исследования пациентов с подозрением на синдром Ангельмана [6]. Сначала необходимо исключить синдром Ангельмана ДНК-анализом. В случае отрицательного результата провести цитогенетический анализ с высоким разрешением полос, затем субтеломерный анализ (чтобы исключить делецию 22q13.3), затем методом сравнительной геномной гибридизации исключить синдромы, сопровождающиеся интерстициальными делециями, и наконец, использовать ДНК-анализ для выявления мутаций в гене МЕСР2.
Если все анализы дали отрицательный результат, необходимо провести полное цитогенетическое и молекулярно-генетическое исследования региона 15q11-q13, исключить дефицит метилен-тетрагидрофолат-редуктазы и мутации в гене АТR-Х [6]. Те же авторы приводят алгоритм обследования больных с мышечной гипотонией и Прадер-Вилли-подобным фенотипом. Этим пациентам необходимо провести ДНК-исследование районов метилирования, исключить синдром FRA-Х, затем осуществить ДНК- анализ на однородительскую дисомию хромосомы 14 и в случае отрицательных результатов провести исследование для выявления МЕСР2 мутаций. Таким образом, чтобы провести полный генетический анализ при подозрении на синдромы Ангельмана или Прадера-Вилли, в некоторых случаях необходимо применить до 8 различных цитогенетических, молекулярно-цитогенетических, молекулярных методов. Пациенты с подозрением на указанные синдромы являются примером многостороннего и целенаправленного исследования, необходимого для правильной постановки диагноза больным [6].
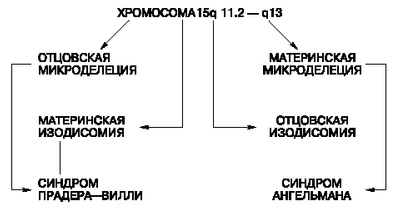
Рис. 5. Возможные механизмы развития синдромов Прадера-Вилли и Ангельмана
В заключение следует еще раз подчеркнуть, что более широкое комплексное применение цитогенетических, молекулярно-цитогенетических методов лабораторного анализа в сочетании с грамотным клиническим обследованием позволит решить проблему диагностики изолированной умственной отсталости или умственной отсталости при наследственных болезнях и синдромах у детей. Это в свою очередь поможет более эффективно осуществлять помощь членам наследственно отягощенной семьи в решении вопросов прогноза потомства и снизить частоту рождения детей с генетическими патологиями.
ГЛАВА 3. СИНДРОМ ДИ ДЖОРДЖИ (ДИ ГЕОРГА)
Синдром Ди Джорджи (Ди Георга, СДД) - изолированный Т-клеточный иммунодефицит. Характеризуется триадой ведущих клинических проявлений: гипоплазия тимуса и/или паращитовидных желез и врожденным пороком сердца. Впервые описан в 1965 году. Поражаются чаще девочки [9, 10, 11].
Наследуется по аутосомно - рецессивному типу [11]. Установлена генетическая характеристика этого врожденного порока развития, в части случаев это аутосомный генетический дефект делеции 22q11.2.
В основе СДД лежит порок развития третьего-четвертого глоточных карманов, возникающий между шестой и десятой неделями гестации, приводящий к агенезии или дисгенезии паращитовидных желез и тимуса. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а заинтересованность пятого кармана проявляется широким спектром врожденных пороков сердца с частым вовлечением дуги аорты [9].
Клиническая характеристика: у большинства больных отмечаются диспластические черты лица. Наиболее характерны диспластичные ушные раковины, гипертелоризм, широкая переносица, "рыбий рот", антимонголоидный разрез глаз. У части детей наблюдаются и более грубые аномалии, такие как микрогнатия и незаращение твердого и мягкого неба. Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям. Гипокальциемические судороги обычно возникают с первых дней жизни. У всех больных отмечается задержка умственного развития. Врожденные пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжелым признакам заболевания [9].
Тимус и паращитовидные железы образуются из третьего и частично четвертого карманов. Паращитовидные железы участвуют в регуляции уровня кальция в крови. Отсутствие данных желез приводит к судорогам вследствие гипокальциемии. Появление судорог у младенца в течение 48 часов после рождения свидетельствует о том, что у ребенка врожденная аплазия тимуса и дефицит Т-клеток. С данным синдромом связаны некоторые другие пороки развития, в том числе расщелина нёба (волчья пасть), щели в носу, низкопосаженные уши и неправильное формирование крупных кровеносных сосудов и сердца. Дети с пороками развития сердечно-сосудистой системы, как правило, умирают. Одно время считали, что этот синдром вызван нарушением нормального эмбрионального развития, которое не обусловлено наследственностью, поскольку не наблюдается семейной предрасположенности к заболеванию и оба пола поражаются одинаково. Впоследствии, однако, было установлено, что многие случаи заболевания связаны со структурными дефектами в 22-й хромосоме [9].
У детей, страдающих синдромом Ди Джорджи, либо совсем нет тимуса, либо он недоразвит, что встречается чаще. Такие дети чрезвычайно восприимчивы к вирусным, бактериальным и грибковым инфекциям и склонны к развитию таких опасных для жизни заболеваний, как цитомегаловирусная инфекция. Иногда в крови ребенка присутствует некоторое количество Т-клеток; такие дети способны сопротивляться инфекциям, при этом с возрастом число Т-клеток постепенно увеличивается [11].
Одна из форм нарушения иммунитета. Проявляется с рождения. Основные симптомы — тетания, обусловленная гипопаратиреозом; повышенная склонность к инфекциям, особенно вирусным и грибковым; аномалии развития костной системы, полости рта, сосудов, внутренних органов. Из инфекционных болезней чаще возникают бронхиты, пневмонии, диарея. Дети отстают и физическом развитии. Болезнь прогрессирует и без лечения к двум годам наступает летальный исход. При исследовании крови определяется низкий уровень, содержании кальции и высокий уровень фосфора, указывающие на паратиреендную недостаточность. Число лимфоцитов в периферической крови обычно нормальное, уровень иммуноглобулинов основных классов также в пределах нормы, но специфический иммунный ответ на введенный антиген снижен. Снижены реакции клеточного типа. Не развиваются и ослаблены реакции замедленной ин нечувствительности. Стимулированный лимфоузел обнаруживает избыточное число фолликулов и плазматических клеток. Наблюдаются уменьшение числа лимфоцитов в паракортикальных тимусзависимых зонах и белой пульпе селезенки, аплазия тимуса, гипоплазия паратиреоидных желез или аплазия их. Аплазия тимуса ведет к нарушению синтеза полноценных иммуноглобулинов. [12].
Иммунологический спектр: количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и NK-клеток и повышенным содержанием В-лимфоцитов. Характерны нормальные или повышенные уровни антител [9].
Показатели гуморального иммунитета у больных с синдромом Ди Джорджа полностью нормальны. Концентрация иммуноглобулинов всех классов нормальная. У некоторых больных повышена концентрация lgE, что, возможно, связано с отсутствием Т-супрессоров. У части больных не удается получить антитела в ответ на иммунизацию [11].
Обязательные лабораторные исследования:
- Анализ крови клинический4
- Тромбоциты 2
- Определение группы крови и резус фактора1
- Анализ мочи2
- Определение уровня кальция в крови2
- Бактериологические исследования содержимого из очагов с определением чувствительности к антибиотикам1
- Общий белок и белковые фракции1
- Трансаминазы АсАТ, АлАТ1
- Маркеры вирусов гепатита В и С в крови1
- С-реактивный белок1
- Сывороточные иммуноглобулины А, М, G1
- Субпопуляции лимфоцитов2
- Концентрация общего IgE в сыворотке1
2. Дополнительные лабораторные исследования:
- Определение уровня паратгормона в плазме1
- Вирусные маркеры (антитела к вирусу Эпштейна-Барр)1
3. Обязательные инструментальные исследования:
Рентгенография органов грудной клетки
УЗИ сердца
ЭКГ
4. Дополнительные инструментальные исследования:
УЗИ органов брюшной полости - 1.
5. Консультации специалистов (по показаниям):
Эндокринолога - 1;
Кардиохирурга - 1;
Отоларинголога - 1;
Окулиста - 1. [7]
Характеристика лечебных мероприятий: иммунная недостаточность, за редким исключением, не определяет прогноз и спектр ведущих клинических проявлений при СДД. В большинстве случаев, если пациент переживает 6-месячный возраст, наблюдается постепенное спонтанное восстановление Т-клеточного иммунитета.
Коррекция Т-клеточных нарушений при СДД может быть достигнута трансплантацией фетального тимуса. При наличии тяжелых пороков, в основном определяющих прогноз для жизни, пересадка тимуса считается недостаточно обоснованной. Лечение пороков сердца ведется по стандартам, принятым в кардиологии, а недостаточности паращитовидных желез - по стандартам эндокринологических отделений [9].
Для лечения синдрома Ди Джорджи с успехом применяют пересадку костного мозга. Кроме того, достигнуты положительные результаты при пересадке эмбрионального тимуса больным с тяжелой степенью поражения [11].
Лечение: трансплантация или имплантация ткани тимуса. Введение тимозина (экспериментально). Противосудорожная терапия, введение препаратов кальция [11].
Длительность стационарного лечения определяется характером и тяжестью клинических проявлений и инфекционных осложнений и варьирует от 3 недель до 3 месяцев [9].
Требования к результатам лечения - купирование клинических проявлений (осложнений) СДД [9, 10].
ВЫВОДЫ
В представленной работе были рассмотрены микроделяционные синдромы: Прадера-Вилли, Ангнльма и Ди Джорджи. Описаны их клинические и генетические проявления, возможные риски. И представлены медицинские методы купирования их проявлений, улучшения состояния больных.
На сегодняшний день синдромы Прадера-Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений - геномного импринтинга и унипарентальной дисомии.
Установлено, что синдром Прадера-Вилли может быть обусловлен двумя основными механизмами. Первый из них - микроделеция хромосомы 15 (15q11.2-q13), которая всегда отцовского происхождения. Второй - материнская изодисомия, т.е. когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера - Вилли обусловлено микроделецией, остальные - дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией.
Для синдрома Ди Джорджи установлено, что он вызывается аутосомным генетическим дефектом делеции 22q11.2. Цитогенетические проявления данного синдрома на сегодня еще не достаточно изучены. Учеными ведется активная работа по решению этой проблемы.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
Гинтер Е.К. Медицинская генетика: Учебник. – М.: Медицина, 2003. – 448с.
Ридли М. Геном. Автобиография вида в 23 главах. – М.: Эксмо, 2008. – 432с.
Денисенкова Е.В., Петрин А.Н., Байков А.Д. Цитогенетические и молекулярно-цитогенетические аспекты диагностики заболеваний, сопровождающихся умственной отсталостью, у детей // Издательство «Медиа Сфера», 2005. Режим доступа:
http://www.mediasphera.ru/journals/detail/206/2988/
Сапиенца К. Геномный импринтинг // В мире науки. (Scientific American. Издание на русском языке). - 1990. - №12. - стр.14-20. Режим доступа: http://www.evolbiol.ru/gen.html
Дуліпа О.Г. Синдром Прадера-Віллі (Prader-Willi Syndrome). Режим доступа: http://www.ibis-birthdefects.org/start/ ... prader.htm
Казанцева Л.З., Новиков П.В., Семячкина А.Н., Николаева Е.А., Курбатов М.Б., Добрыкина Э.В. Московский НИИ педиатрии и детской хирургии Минздрава РФ. Синдром Прадера - Вилли у детей: новое в этиологии, патогенезе и лечении. Режим доступа: http://nature.web.ru/db/msg.html?mid=1174772&uri=index2.html
Миронов М.Б., Мухин К.Ю., Кузина Н.Ю., Боровиков К.С., Гоева И.А., Красильщикова Т.М., Коркин А.В., Петрухин А.С. Синдром Ангельмана. Клинический случай. Режим доступа: http://www.epileptologist.ru/pub/Angelman.html
Белозеров Ю.В., Леонтьева И.В., Школьникова М. А., Страхова О.С., Себелева И.А., Макаров Л.М., Давыдкин В.В., Динов Б.А. Наследственные болезни сердца у детей // Мир науки и культуры, 2000-2009. Режим доступа: http://nature.web.ru/db/msg.html?mid=1166264&s=111400060
Соколов В.Н. Гипопаратериоз // Наука и практика, 2009. Режим доступа: http://www.chtfoms.ru/index.php?option=com_content&task=view&id=26467&Itemid=24
Ярцев М.Н., Яковлева К.П., Плахтиенко М.В. ГНЦ Института иммунологии ФМБА России, Москва Иммунодефицитные состояния у детей // Издательство Media Medica, 2000. Режим доступа: http://old.consilium-medicum.com/media/pediatr/06_01/4.shtml
Вилочковой железы аплазия врожденная (Ди Джорджа синдром). Режим доступа: http://nasledbolezn.org.ua/obmen-veshestv/didjordja-sindrom.html
ПРИЛОЖЕНИЕ
Рис. 1. 15 хромосома


Рис. 2. Делеция 15 q11 – 13.1

Рис. 3. Дети с синдромом Прадера-Вилли

Рис. 4. Мальчик с синдромом Прадера-Вилли
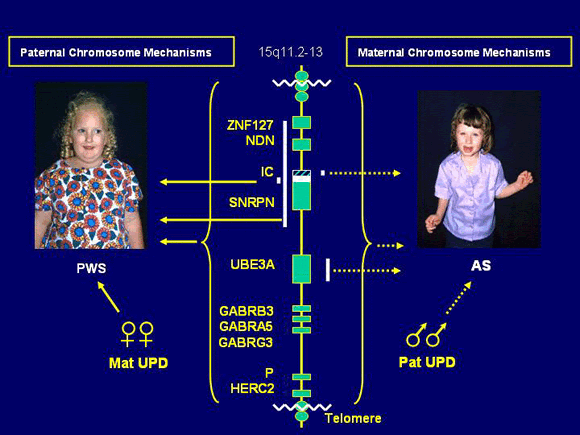
Рис. 5. Синдромы Прадера-Вилли и Ангельмана

Рис. 6. Мальчик с синдромом Ангельмана
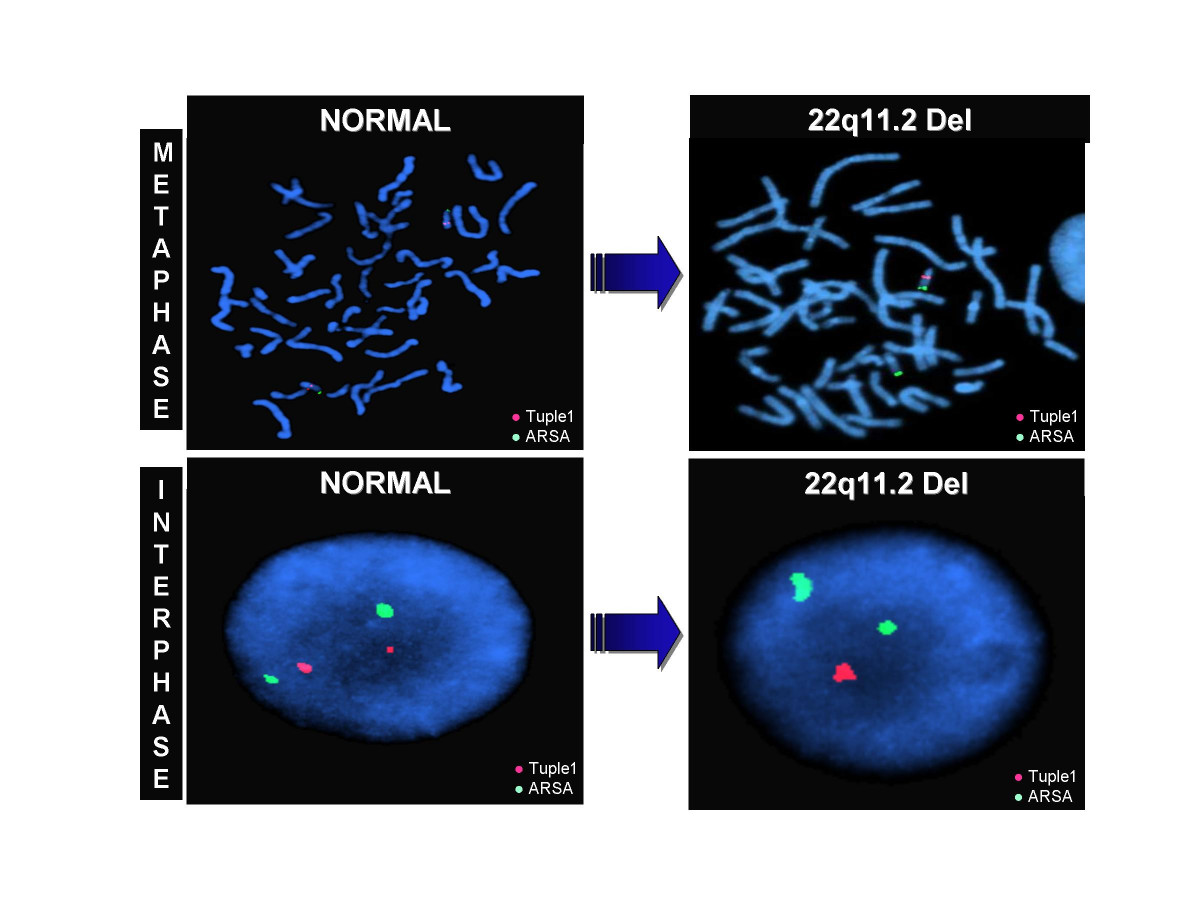
Рис. 7. Делеция 22q11.2 – синдром Ди Джорджи
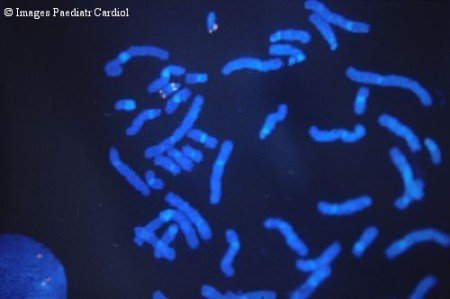
Рис. 8. Делеция 22q11.2 – синдром Ди Джорджи
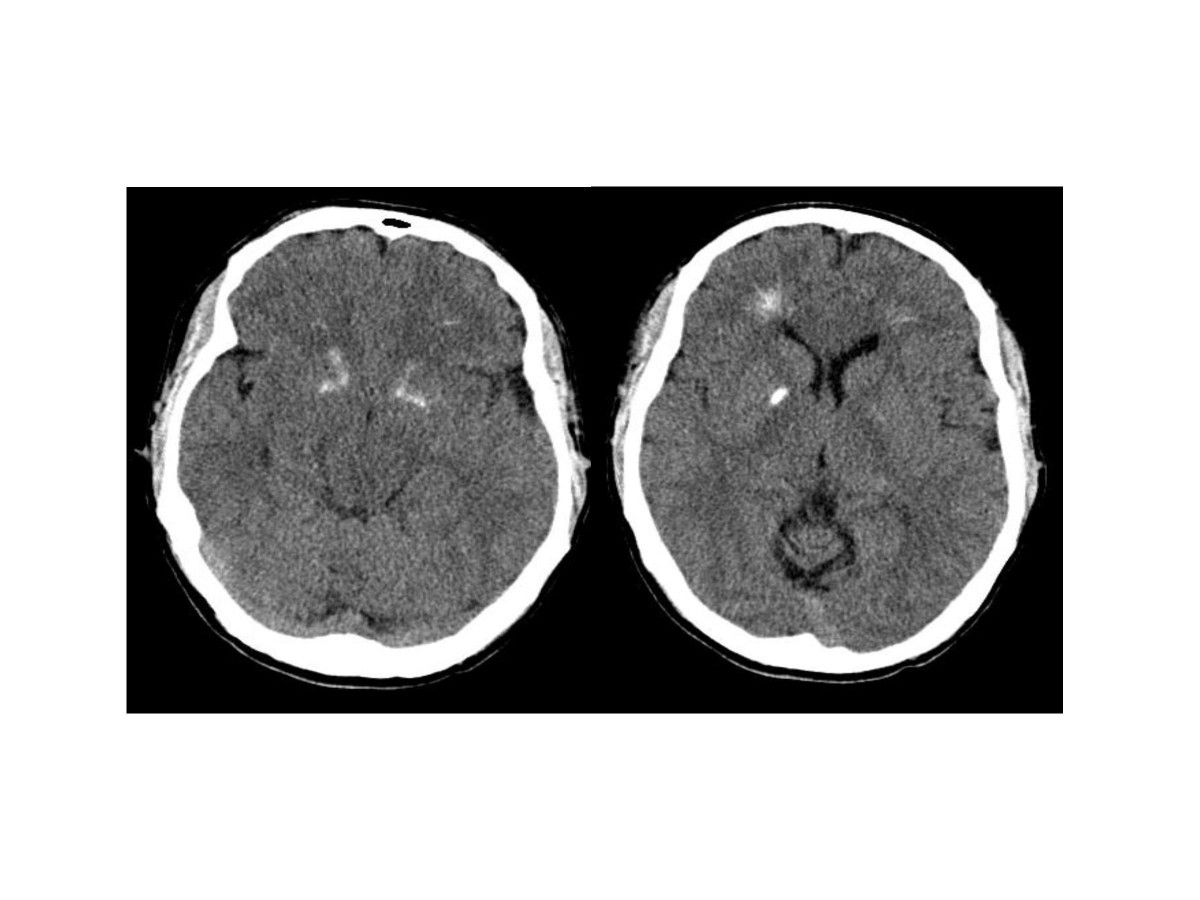
Рис. 9 Компьютерная томограмма мозга пациента с делецией
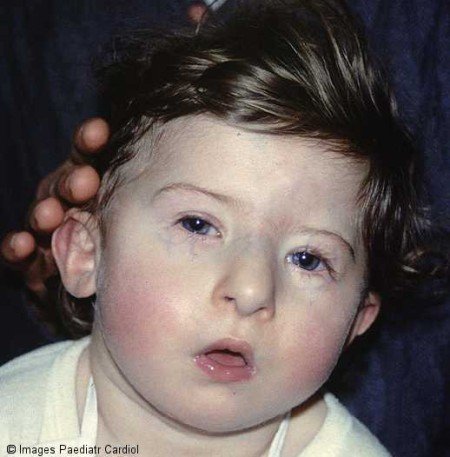
Рис. 10. Синдром Ди Джорджи

Рис. 11. Девочка с синдромом Ди Джорджи

Рис. 12 Мальчик с синдромом Ди Джорджи