Разработка ионометрической методики
Разработка ионометрической методики
Процесс разработки ионометрической методики интересен не только исследователям, разрабатывающим методики, но более широкой аудитории химиков, которая занимается только эксплуатации этих методик. На первый взгляд это утверждение несколько спорно, но не будем спешить с выводами. Опыт показывает, что опытный химик-аналитик, эксплуатирующий методику, всегда ясно представляет себе необходимость того или иного этапа методики, целесообразность присутствия того или иного реактива в анализируемой смеси. Вершиной аналитического мастерства является формирование критического взгляда на внедряемую в эксплуатацию методику еще на стадии прочтения текста методики. В полной мере владеть этим мастерством можно только в том случае, если владеть наиболее полной информацией о процессе разработки методики.
Рассмотрим типичные стадии процесса разработки ионометрической методики.
Разработка любой ионометрической методики, как и любой другой аналитической, начинается с определения потенциального состава анализируемой пробы. Прежде всего, следует обладать информацией о возможном диапазоне концентраций определяемого вещества, наличии сопутствующих примесей, кислотной реакции среды пробы.
Второй этап разработки методики состоит в выборе электрода, который бы позволил бы проводить ионометрические измерения в выбранном диапазоне концентраций в присутствии мешающих анализу примесей.
Во-первых, рабочий интервал концентраций ионоселективого электрода (ИСЭ), который указывается в паспорте электрода, должен быть больше предполагаемого диапазона концентраций определяемого вещества пробы.
Во-вторых, примеси, входящие в состав пробы, не должны оказывать сильного влияния на результат анализа. Действие примесей может проявляться двояко. Прежде всего, примеси могут образовывать с анализируемым ионом прочные соединения, занижая, таким образом, результаты анализа. Такое положение дел характерно не только для ионометрических определений, и поэтому решать эту задачу следует способами, которые подсказывает богатый опыт общей аналитической химии. Примеси также могут влиять на результаты анализа, искажая аналитический сигнал ионоселективного электрода. В этом случае несложно произвести расчет погрешности определения от влияния мешающих анализу примесей. Относительная погрешность анализа от действия каждой примеси можно рассчитать по следующей формуле:
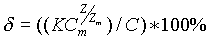
C - концентрация определяемого иона;
Z - заряд определяемого иона;
C>m> - концентрация мешающего иона;
Z>m> - заряд мешающего иона;
K - коэффициент селективности. (Коэффициенты селективности важных мешающих ионов обычно указываются в паспортах, прилагаемых к ИСЭ.)
Если вычисленная относительная погрешность слишком высока, то следует позаботиться о процедуре маскирования той или иной мешающей примеси.
На пути предварительных расчетов есть свои осложнения. Главная сложность состоит в том, что список мешающих ионов, что указывается в паспорте на электрод, недостаточно велик. Как поступать в этом случае?
Во-первых, следует поискать подобные электроды, так как есть большая вероятность того, что электродноактивный материал электрода давно известен и хорошо изучен. За многообразием торговых названий ионоселективных электродов известных отечественных фирм скрывается довольно ограниченный список действительно новых электродов. В связи с этим есть смысл поинтересоваться у производителей составом материала электрода. Ответ не всегда можно получить, но поинтересоваться стоит. Если поиски свойств электрода ни к чему не привели, то ничего не остается делать, как проводить собственные исследования.
Итогом всех расчетов второго этапа должен быть вывод о пригодности выбранного электрода для решения вашей аналитической задачи.
Третий этап разработки ионометрической методики состоит в выборе метода анализа. При выборе метода анализа надо иметь в виду, что прямая потенциометрия привлекательна, главным образом, несложной процедурой анализа, метод добавок - надежностью результатов, а титриметрия - их точностью.
Четвертый этап целиком посвящается расчету состава раствора, регулирующего ионную силу и кислотность среды, маскирующего влияние мешающих компонентов.
Регулировка ионной силы раствора производится солями электролитов. Электролит добавляется для того, чтобы ионную силу в растворе определял он, а не компоненты анализируемого раствора. В связи с этим снижается ошибка от неконтролируемого изменения активности анализируемого иона. Нет нужды говорить о том, что компоненты электролита не должны влиять на отклик ионоселективного электрода. Расчет концентрации электролита можно производить с помощью программы IonCalc.
Для регулирования кислотности среды пробы используются традиционные в аналитической химии буферные смеси. Также как и электролит, вещество буфера не должно влиять на аналитический сигнал ИСЭ.
Удачным считается тот приготовленный раствор, который содержит как можно меньше компонентов. Примером может служить раствор для анализа фторидов фторидселективным электродом. Раствор содержит лимонную кислоту, нейтрализованную едким натром до pH=5-6. Эта смесь выполняет одновременно роль электролита для создания ионной силы, реагента для поддержания необходимой кислотности среды и маскирующего реагента. Функция смеси, как маскирующего реагента, состоит в разрушении комплексов фторида с катионами железа и алюминия, которые могут присутствовать в пробе.
Применим полученные знания на примере разработки методики для определения хлоридов в речной воде. Известно, что в речной воде содержатся следующие ионы:
|
Ca2+ |
10-3 - 10-2 M |
|
HCO>3>- |
2 10-3 - 2 10-2 M |
|
Cl- |
5 10-5 - 5 10-4 M |
|
Na+ |
10-5 - 10-4 M |
|
pH |
7 -9 |
Содержание хлорида в речной воде указывает на то, что исходя из предела обнаружения, можно использовать электрод типа ЭМ-Сl-01. Рабочий диапазон этого электрода составляет 310-5 - 1М при pH=2-11. Паспортные данные о мешающих ионах говорит о том, что примеси нашего анализируемого объекта практически не оказывают никакого влияния на определение (см. Ионоселективные электроды). Единственным фактором, снижающим точность определения, является высокое содержание ионов кальция и гидрокарбоната. Колебание концентрации этих ионов будет существенно изменять активность ионов хлора. Есть 2 выхода из этого положения: добавка электролита большой концентрации или использование для анализа метода добавок, а не прямой потенциометрии. (В методе добавок колебание ионной силы раствора меньше сказывается на результатах анализа.) Если останавливаться на прямой потенциометрии, то в качестве электролита, поддерживающего ионную силу раствора, можно использовать нитрат калия квалификации "х.ч" или "ч.д.а.". Квалификация реактива важна постольку, поскольку в реактиве содержится хлорид. Нормируемая величина хлорида в реактиве "х.ч." составляет 5 10-4%.
Расчеты с помощью программы IonCalc показывают, что если использовать 0,1М раствор нитрата калия, то систематическая ошибка определения от разницы между ионной силой калибровочных растворов и ионной силой анализируемого раствора может составить 2,8%. Примеси хлорида в нитрате калия незначимы, поскольку составляют 5 10-7М в растворе.
В заключении следует сказать о том, что применять буферы кислотности среды для этой методики не рационально, так как колебание pH в пробах невелико.
Список литературы
Для подготовки данной применялись материалы сети Интернет из общего доступа